










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkDYNA
Print version ISSN 0012-7353
Dyna rev.fac.nac.minas vol.82 no.189 Medellín Jan./Feb. 2015
https://doi.org/10.15446/dyna.v82n189.42131
http://dx.doi.org/10.15446/dyna.v82n189.42131
Kinetic aspects on ferric arsenate formation in a fix bed gas-solid reaction system
Aspectos cinéticos de la formación de arseniato férrico en lecho fijo mediante reacciones gas-sólido
Eduardo Balladares a, Roberto Parra b & Mario Sánchez c
a Departamento de Ingeniería Metalúrgica, Universidad de Concepción, Concepción, Chile. eballada@udec.cl
a Departamento de Ingeniería Metalúrgica, Universidad de Concepción, Concepción, Chile. rparra@udec.cl
b Facultad de Ingeniería, Universidad Andrés Bello, Santiago de Chile, Chile. mario.sanchez@unab.cl
Received: February 18th, 2014. Received in revised form: August 20th, 2014. Accepted: August 22th, 2014.
Abstract
The fixation of arsenic contained in gases produced during pyrometallurgical processes by using solid ferric oxide was studied in the range 873-1073 K under different oxygen potential and solid aggregates porosities. Arsenic fixation on solid iron oxides is described by the pore blocking model under the studied conditions. The solid product of the reaction has a molar volume 3 times larger than the solid reactant causing fast decreasing of the inter-granular spacing. The activation energies of arsenic fixation reaction are 34.96 and 35.46 kJ/mol for porosities of 0.88 and 0.74 respectively, and for porosity of 0.55 the activation energy was 26.88 kJ/mol. These values of activation energy show that intra-pellets diffusion has an effect only in samples with 0.55 porosity. Minor sintering of particles was detected. Industrial application of the concept demands a reaction system, which in is required better gas-solid contact for attaining larger conversions.
Keywords: arsenic fixation; flue dust; gas treatment; copper smelting.
Resumen
Se presentan los resultados para la fijación de arsénico contenido en una corriente gaseosa en el rango 873-1073 K, bajo diferentes presiones parciales de oxígeno en lechos de distinta porosidad. Se estudió la cinética de formación de arseniato férrico y se determinó que la reacción puede describirse por el modelo de bloqueo de poros, verificándose un aumento del volumen molar del producto formado limitando la difusión de arsénico gaseoso hasta la interfase de reacción. Las energías de activación de la reacción 0.25As4O6(g)+0.5O2(g)+0.5Fe2O3(s)=FeAsO4(s) fueron 34.96, 35.46 y 26.88 kJ/mol para las porosidades 0.88, 0.74 y 0.55, respectivamente. Estos valores de energía de activación muestran que la difusión intra-pellet juega un rol importante solo para porosidades de lecho bajas (e=0.55). Se evidenció un incipiente grado de sinterización de las partículas. Lograr contacto más íntimo gas-partícula para mayores retenciones de arsénico resulta clave para su aplicación en la industria pirometalúrgica del cobre.
Palabras clave: fijación de arsénico; gases de fundición; tratamiento de gases; fundición de cobre.
1. Introducción
En la actualidad, la mayor parte de los concentrados de cobre en Chile son procesados por métodos pirometalúrgicos, generando enormes caudales de gases que contienen, entre otros elementos tóxicos, arsénico. Este elemento causa problemas tanto desde la perspectiva ambiental como operacional debido a las crecientes emisiones, por una parte, y a los requerimientos de calidad de ánodos y ácido, por otra. Así, la remoción de arsénico desde la fase gas para producir un producto sólido estable disminuiría los problemas causados por el manejo de ingentes volúmenes de solución cuando estos se tratan por métodos hidrometalúrgicos convencionales. La fijación de arsénico con óxido de hierro (III) es termodinámicamente más favorable que con otros óxidos [1]. De este modo, la formación de arseniato férrico desde una corriente con trióxido de arsénico gaseoso con Fe2O3 sólido se presenta como muy atractiva debido a la estabilidad térmica y acuosa del arseniato férrico.
La Fig. 1 muestra un diagrama de predominancia de áreas para el sistema Fe-As-S-O en las condiciones en las que los gases salen del horno de fusión. En este se observa que la única fase estable portadora de arsénico es el trióxido de arsénico gaseoso (As2O3(g) dimerizado o As4O6(g)). Por otra parte, dado que los potenciales de S2 (o equivalentemente de SO2) en los gases de fusión están en el rango pS2=10-1-10-3 atm, el hierro es estable en la forma de sulfato férrico, tal como lo muestra el diagrama de predominancia de áreas de la Fig. 1. La reacción de formación de FeAsO4 a partir de As4O6 y FeSO4 tiene gradiente termodinámica negativa a 1573 K (DGº(1573 K)= -142 kJ/mol de FeAsO4) [2] y requiere de cantidades extremadamente bajas de As en fase gas (pAs4O6=10-22 atm a 1573 K), no obstante, en la práctica no se ha detectado cantidades significativas de FeAsO4 en las caracterizaciones de los polvos de fundición captados desde los sistemas de limpieza y manejo de gases [3]. De aquí, se plantea la opción de fijar el arsénico (o parte de él) en una etapa adicional mediante una reacción heterogénea en la que la reacción global, descrita por la reacción (1), idealmente podría dar cuenta de una fracción significativa del arsénico en una fase estable.
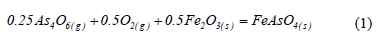
Los antecedentes recién mencionados, así como completos análisis termodinámicos previamente reportados ampliamente en la literatura [4-8], confirman la factibilidad de fijar el arsénico como FeAsO4 de acuerdo a la reacción (1). De este modo, el presente trabajo está dedicado al análisis cinético del proceso.
2. Experimental
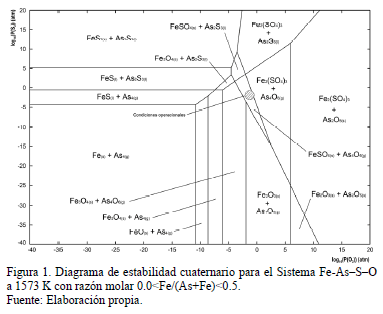
Los experimentos se realizaron en una balanza termogravimétrica tal como muestra la Fig. 2. En esta, el arsénico fue producido por volatilización de As2O3 sólido de 99.2 % de pureza y Fe2O3 de 99% pureza, esta última en forma de polvo fino (100% -300 mm), ambos reactivos proporcionados por Riedel de Haën. Las experiencias se efectuaron en un horno vertical eléctrico que contenía el reactor para producir el As4O6(g) y el posterior contacto de este gas con la hematita sólida fue en un tubo de cuarzo de 18 cm de diámetro. Las muestras fueron ubicadas sobre un contenedor de óxido de aluminio poroso de modo tal que el gas circulara a través suyo. Este contenedor pendía de una balanza y estaba protegido por una campana de cuarzo cuyo detalle se muestra en la Fig. 3. En una prueba típica, la muestra se suspendía en la balanza termogravimétrica dentro del horno eléctrico. El flujo de gas se alimentaba desde la parte inferior del horno y la zona de reacción estaba ubicada en la sección isotérmica del horno. Los reactivos fueron hematita en polvo, como fuente de hierro, y un flujo de gas, constituido por O2(g) y As4O6(g), el que proporcionaba arsénico y oxígeno necesarios para la reacción. Se empleó un flujo de nitrógeno como gas de arrastre para el As4O6, el que volatilizaba en el fondo del tubo de reacción dentro del horno. El oxígeno fue alimentado por medio de un tubo de ¼" de diámetro dentro de un contenedor de cuarzo poroso, para las experiencias que utilizaron un lecho de hematita sólida, dentro de un cesto especialmente diseñado para las experiencias con pellets.
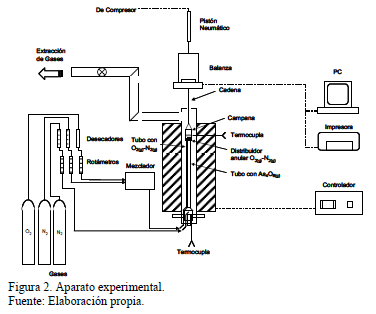
En la parte inferior del horno, donde el As4O6 es volatilizado, la temperatura fue mantenida constante a 573 K mediante un controlador conectado a una termocupla tipo K,
asegurando una tasa de volatilización constante. La balanza termogravimétrica fue conectada a un computador para la adquisición de datos en línea de la evolución del peso de la muestra. El peso instantáneo se desplegó en pantalla y se almacenó en un archivo de datos. Los gases producidos durante los experimentos fueron evacuados desde la parte superior del horno empleando un extractor. Este extractor mantenía una presión interna en el reactor levemente negativa permitiendo una pequeña infiltración en la sección superior del reactor, después de la zona de reacción, evitando emisiones de gases arsenicales al exterior.
Con el objetivo de mantener el flujo de gas arsenical constante en la zona de reacción (parte superior del reactor), se realizó pruebas de volatilización de As2O3 a diferentes temperaturas obteniéndose las correspondientes tasas de volatilización. Sin embargo, a temperaturas superiores a 603 K la tasa de volatilización resulta demasiado alta y, de este modo, se requería una cantidad de As2O3 mayor a la capacidad del receptáculo para el tiempo de duración de las pruebas. Por otra parte, a temperaturas inferiores a 523-553 K, es difícil mantener la temperatura de la zona de volatilización constante y, por tanto, también la temperatura de la zona de volatilización. De este modo, se seleccionó 573 K como la temperatura de volatilización ya que a esta se logró una tasa de volatilización constante y estable.
3. Resultados y discusiones
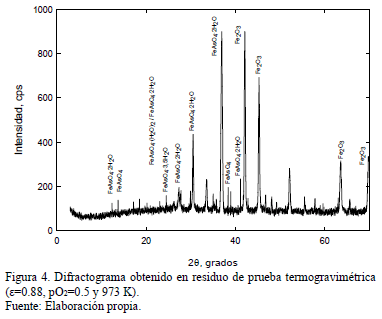
La caracterización inicial realizada a los productos de reacción mostró la presencia de ciertas cantidades de compuestos del tipo FexAsyOz, tal como se muestra en el espectro de difracción de rayos X de la Fig. 4, obtenido para e=0.88, pO2=0.5 y 973 K. En esta, se comprueba la presencia de FeAsO4 además de FeAsO4•2H2O, FeAsO4•(H2O)2, FeAsO4•3.5H2O y, Fe2O3 no reaccionado. Dada la ausencia de H2O en el sistema de reacción, la presencia de arseniato hidratado se explica en el manejo de las muestras post-reacción, favorecido por el comportamiento higroscópico del FeAsO4.
La evaluación de los resultados se basó en la determinación de la conversión (o fracción convertida) de Fe2O3 a FeAsO4. La conversión total (registrada una vez que la prueba finaliza) se estimó mediante la expresión:
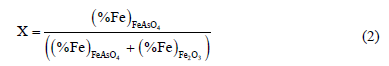
mientras que la fracción convertida instantánea está dada por:

donde (%Fe)FeAsO4 y (%Fe)Fe2O3 representan el contenido de hierro en la forma de FeAsO4 y Fe2O3, respectivamente. Mientras que Wt, Wi y W¥ representan el peso de la muestra en el tiempo t, el peso inicial y el peso para una conversión completa, respectivamente.
3.1. Efecto de la temperatura
Pruebas preliminares mostraron que la reacción se detenía a aproximadamente 80 minutos de iniciada, sin obtenerse información adicional después de este tiempo. De este modo, este tiempo fue seleccionado como tiempo de reacción fijo para todos los ensayos realizados.
El efecto de la temperatura fue estudiado en el rango 873-1073 K. La Fig. 5 muestra el efecto de la temperatura sobre la conversión de Fe2O3 en FeAsO4 donde se observa que la conversión aumenta progresivamente hasta 1073 K.
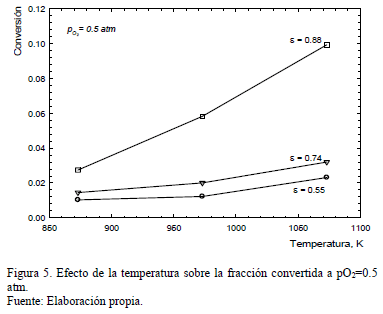
No obstante, en las pruebas en las que se empleó pellets (e=0.55 y 0.74) el efecto de la temperatura fue marginal, aumentando la conversión solo levemente. En cambio, en las pruebas en las que se empleó Fe2O3 en lecho fijo (e=0.88) el aumento en la conversión registrado fue significativo (se cuadruplicó al aumentar la temperatura de 873 a 1073 K).
Aun cuando el efecto de la temperatura es mucho más marcado en reacciones controladas por la cinética química, comparado con aquellas controladas por transporte en el fluido o difusión gaseosa, en este caso, el aumento de la conversión no es exponencial con la temperatura por lo que no se puede inferir que el control sea químico.
3.2. Efecto de la porosidad
El efecto del área superficial sobre la extensión de la conversión fue estudiado mediante experiencias en lecho poroso, cuyos resultados se muestran en la Fig. 6 para una presión parcial de oxígeno de 0.5 atm. A 1073 K la conversión obtenida en muestras con porosidad e=0.88 aumenta 5 veces respecto a aquella alcanzada a e=0.55. Para muestras compactas, tales como pellets (con e=0.55), la reacción se inicia principalmente sobre la superficie exterior del pellet, de modo que a medida que la reacción avanza los poros se obstruyen y la reacción se detiene progresivamente, lográndose conversiones inferiores en comparación con muestras más porosas (polvo de Fe2O3 en lecho fijo). Esto puede ser explicado al comparar los volúmenes molares de los reactantes y productos sólidos. Específicamente, el FeAsO4 posee un volumen molar 3 veces superior al Fe2O3, así, por cada cm3 de Fe2O3 sólido reaccionado se forman 3 cm3 de FeAsO4, donde el aumento del volumen molar está dado por la expresión (4).

donde PMP y PMR son los pesos moleculares de producto y reactivo, rP y rR las densidades de producto y reactivo y a es la razón de coeficientes estequiométricos en la reacción (1), en este caso = ½.
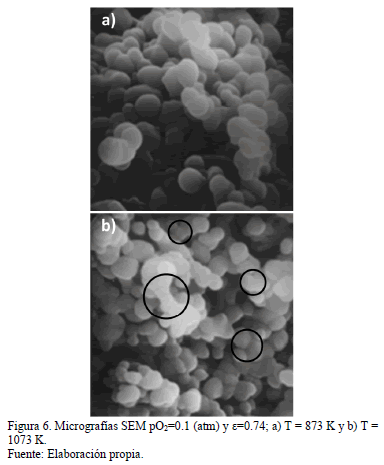
De lo anterior, se registra una reducción gradual del área superficial debido al bloqueo de poros externos, disminuyendo así la superficie inter-partícula. Este hecho coincide con resultados previamente reportados [9-12].
La Fig. 6 (a) y (b) muestra leves diferencias en las formas de los límites inter-partícula, los que se señalan en círculos negros. En el caso de la muestra reaccionada a 1073 K (derecha) los bordes en contacto entre las partículas son mucho más paralelos, observándose, inclusive, granos prácticamente soldados. Aun cuando 1073 K es una temperatura baja comparada con la temperatura de fusión, tal evidencia podría indicar la posibilidad de sinterización localizada, más aun considerando que la reacción de formación del FeAsO4(s) dada por la reacción (1) es exotérmica, tal como lo indica su calor de reacción [13]:
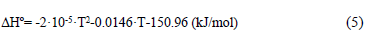
La expresión anterior muestra que la reacción es exotérmica en todo el rango de temperatura de interés, más aún, los datos indican que varía muy poco en el rango 873-1073 K, específicamente, de -151.6 a -150 (kJ/mol). En efecto, la generación de calor en la interfase de reacción (y la posible resistencia a que este fuera disipado) podría hacer aumentar la temperatura localmente, favoreciendo el mecanismo antes mencionado.
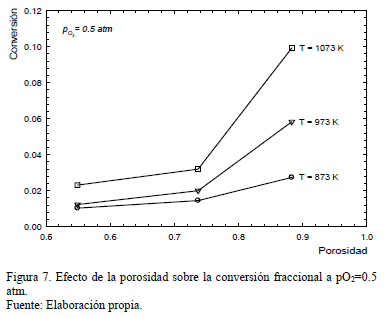
La Fig. 7 muestra el efecto de la porosidad sobre la conversión de Fe2O3 a FeAsO4 a pO2= 0.5 atm a las 3 temperaturas evaluadas. En esta se observa claramente que muestras más porosas de sustrato hematítico producen mayores conversiones dada la mayor superficie de reacción disponible. El efecto es más marcado cuanto mayor es la temperatura dando cuenta de lo relevante de la disponibilidad de interfase de reacción en la cinética global, medida como conversión en la Fig. 7.
3.3. Cinética global de reacción
La disminución de la superficie inter-partícula debido al bloqueo de poros se verificó por medio de microscopía electrónica de barrido (SEM), la cual muestra una estructura de poros de las muestras más compactas una vez finalizada la reacción en comparación con las muestras en su estado inicial. La evidencia experimental obtenida para la formación de arseniato férrico a partir de Fe2O3 y arsénico contenido en fase gas sugiere que el proceso es representado más apropiadamente por el modelo de bloqueo de poros que empleando otros modelos, tales como: cinética de primer o segundo orden, modelos de nucleación y crecimiento y ecuaciones de núcleo recesivo, entre otros. Así, de acuerdo a la ecuación de Evans [14], la expresión para la tasa de reacción con bloqueo de poros está dada por la ecuación:

donde l es el parámetro de bloqueo de poros, k' es la constante de velocidad aparente y t es el tiempo de reacción. La constante de velocidad está dada en términos de la temperatura y la presión parcial de oxígeno de acuerdo a:

donde b es el coeficiente estequiométrico de la reacción (1), k es la constante cinética química y f(pO2) es una función de la presión parcial de oxígeno.
Aunque la temperatura y el grado de obstrucción de poros tienen un leve efecto sobre l, los datos experimentales dan un valor en torno a 0.05, por lo que este puede ser considerado constante como una primera aproximación para ajustar el modelo. Además, se verificó que el parámetro l es casi independiente de la presión parcial de oxígeno.
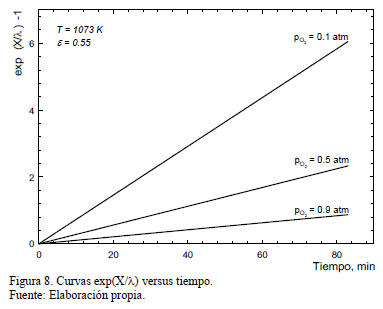
La constante de velocidad aparente, k', se obtiene de las pendientes de las curvas de la Fig. 8 y estos valores son entonces graficados como una función de pO2 en la Fig. 9 para ilustrar el efecto de la presión parcial de oxígeno sobre k'. En esta Figura se muestra también los resultados obtenidos en los experimentos que emplearon muestras menos porosas (e=0.55). Los resultados obtenidos para porosidades mayores (e=0.88 y e=0.74) mostraron igual comportamiento.
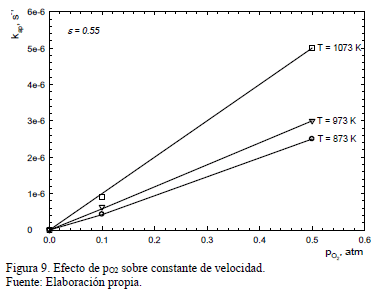
Los resultados experimentales obtenidos tanto para las porosidades mayores (e=0.88 y e=0.74) como para aquellas obtenidas de los pellets (e=0.55) indican que la reacción es de primer orden con respecto a la presión parcial de oxígeno. Po lo tanto, la expresión (7) puede re-escribirse de la forma:

Esta ecuación permite obtener las constantes de velocidad, k, desde las pendientes de las líneas obtenidas al graficar k' versus pO2, y los valores obtenidos se muestran en la Tabla 1.
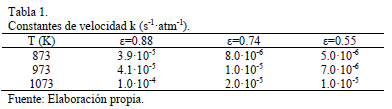
Consecuentemente, la energía de activación se determina graficando estos valores en curvas tipo Arrhenius. Así, los resultados para las tres porosidades probadas se muestran en la Fig. 10.
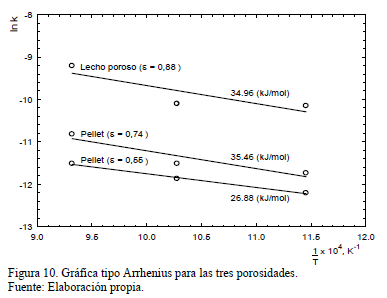
Las energías de activación obtenidas para el lecho poroso, así como aquellas para pellets, muestran que la reacción tiene lugar bajo control difusional. Los valores obtenidos para lecho poroso (e=0.88) y pellet con e=0.74 son muy similares, lo que sugiere la ausencia de efecto de difusión intra-pellet. Sin embargo, para pellets con porosidad e=0.55 la energía de activación obtenida indica que los efectos difusionales intra-pellet son significativos y afectan la velocidad de reacción global. La Tabla 2 muestra un resumen de los parámetros cinéticos obtenidos para todas las condiciones estudiadas en el laboratorio.

4 Conclusiones
Los resultados experimentales obtenidos de las pruebas termogravimétricas muestran que es posible fijar arsénico empleando óxido férrico como sustrato, aunque las conversiones obtenidas son relativamente bajas.
Para lograr mejores retenciones de arsénico se debe diseñar e implementar un sistema experimental que proporciones un mayor contacto gas-sólido. Dado que se verificó cierto grado de sinterización en el interior de las muestras parcialmente reaccionadas, mayores temperaturas no serían de utilidad para lograr este objetivo pues se incrementaría tal sinterización.
Los valores de energía de activación obtenidos muestran que la difusión intra-pellet no afecta a la tasa global de reacción, excepto para el caso de las muestras de menor porosidad, esto porque probablemente en las muestras menos porosas la superficie se bloquea muy rápidamente.
Agradecimientos
Los autores desean expresar su gratitud al apoyo financiero de CONICYT por los fondos provenientes del Proyecto de Investigación Científica Nº1000474.
Referencias
[1] Ulloa, A., Sánchez M. and Terry, B., Lime roasting of enargite, Proceedings of Pyrometalurgy '95, IMM Cambridge, pp. 275-283, July 1995. [ Links ]
[2] Barin, I., Thermochemical data of pure substances, Part I, VCH Verlags Gesellschaft, Weinheim, 1993. [ Links ]
[3] Balladares, E., Tesis de MSc., Departamento de Ingeniería Metalúrgica, Universidad de Concepción, Concepción, Chile, 2004. [ Links ]
[4] Lynch, D C., A review of the physical chemistry of arsenic as it pertains to primary metals production. Arsenic Metallurgy Fundamentals and Applications, Ed. by R. Reddy, J. Hendrix and P. Queneau, TMS Inc., pp. 3-33, 1987. [ Links ]
[5] Mah, A.D., Thermodynamic data for arsenic sulfide reactions. U.S. Department of Interior, Bureau of Mines, Report of Investigations Nº8671, pp. 1-85, 1982. [ Links ]
[6] Vian, A., Iriarte, C. and Romero, A., Fluidized roasting of arsenopyrites. I. & E. C. Process Design and Development, 2 (3), pp. 214-223, 1963. http://dx.doi.org/10.1021/i260007a008 [ Links ]
[7] Shigematsu, K., Vapor pressure measurements of arsenic compounds. Metallurgical Review of MMIJ, 3 (2), pp. 29-48, 1986. [ Links ]
[8] Skeaff, J. M., Thermochemical arsenate-oxide equilibria in the system Fe-As-O determined electrochemically between 317 and 536 ºC. Canadian Metallurgical Quarterly, 28 (2), pp. 117-126, 1989. http://dx.doi.org/10.1179/cmq.1989.28.2.117 [ Links ]
[9] Chaubal, P.C. and Sohn, H.Y., Intrinsic kinetics of the oxidation of chalcopyrite particles under isothermal and nonisothermal conditions. Metallurgical Transactions, 17B, pp. 51-60, 1986. http://dx.doi.org/10.1007/BF02670818 [ Links ]
[10] Wang, G.X., Chandra, D. and Fuerstenau, M.C., Oxidation of cobaltite: Part II. Kinetics. Metallurgical Transactions, 26B, pp. 523-527, 1995. http://dx.doi.org/10.1007/BF02653869 [ Links ]
[11] Björkman, B., Degerstedt, U., Lindblom, B. and Samuelsson, C., Kinetics of impurity elimination during roasting. In Extraction and Processing for the Treatment and Minimization of Wastes, Ed. by J. Hager, B. Hansen, W. Imrie, J. Pusatori and V. Ramachandran, The Minerals, Metals & Material Society, pp. 825-842, 1993. [ Links ]
[12] Fan, Y.H., Tesis de MSc., Departamento de Ingeniería Metalúrgica, Universidad de Concepción, Concepción, Chile, 1997. [ Links ]
[13] Ronie, A., et al., Outokumpu Research Oy, 2006. [ Links ]
[14] Evans, U.R., The corrosion and oxidation of metals: Scientific principles and practical applications. Ed. Edward Arnold Ltd., 1960. [ Links ]
E. Balladares, se tituló de Ingeniero Metalúrgico en 1994, así como los grados de MSc. En 2004 y Dr. En 2008 en Ingeniería Metalúrgica en la Universidad de Concepción, Chile. De 1995 a 2001 trabajó como Ingeniero de procesos, Jefe de Planta y Jefe de Producción en SQM Salar. De 2002 a 2008 trabajó como Ingeniero de Proyectos y Consultor. Desde 2008 es Profesor Asistente en el Departamento de Ingeniería Metalúrgica de la Universidad de Concepción, Chile.
R. Parra, titulado de Ingeniería Metalúrgica en 1991 de la Universidad de Concepción, C hile, obtuvo un D.E.A. en 1992 y un PhD. En 1998, en Ciencia e Ingeniería de Materiales, ambos en el Institut National Polytechnique de Grenoble, France. Profesor titular en el Departamento de Ingeniería Metalúrgica de la Universidad de Concepción, Chile. Sus principales áreas de interés en I+D son la Fisicoquímica de procesos a alta temperatura con especial énfasis en la pirometalurgia del cobre. Es Profesor Adjunto en la Escuela de Minas de Oviedo, España, donde desarrolla actividades académicas en el grupo de Investigación de Siderurgia, Metalurgia y Materiales. Es también investigador asociado del grupo de Sustentabilidad en procesos siderúrgicos y metalúrgicos en el CENIM, Madrid, España.
M. Sánchez, es Ingeniero Civil Metalúrgico de la Universidad de Concepción, Chile. Dr. Ingeniero del Instituto Nacional Politécnico de Grenoble, Francia. PhD. en el Imperial College de Londres. Actualmente Director y Profesor de la Carrera de Ingeniería Civil en Metalurgia de la Universidad Andrés Bello, Santiago, Chile. Se ha desempeñado como docente y/o investigador en seis Universidades de América y Europa. Ha ejercido actividades profesionales en Fundiciones de Potrerillos y Chuquicamata de Codelco y en Hatch Ingenieros, Chile. Es autor de tres textos sobre Pirometalurgia del cobre, editor de 8 proceedings en el campo del Medio Ambiente en Minería y Metalurgia y editor de los proceedings de la VIII Conferencia Internacional Molten 2009. Es autor o co-autor de cerca de cien artículos científicos y técnicos en revistas y conferencias.
