










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkDYNA
versión impresa ISSN 0012-7353
Dyna rev.fac.nac.minas vol.83 no.196 Medellín mar./abr. 2016
https://doi.org/10.15446/dyna.v83n196.51933
DOI: http://dx.doi.org/10.15446/dyna.v83n196.51933
Synthesis and modification of beta zeolite for use in toluene disproportionation reaction
Síntesis y modificación de la zeolita beta para su uso en la reacción de desproporción de tolueno
Santiago Mesa a, Johana Arboleda b & Adriana Echavarría c
a Instituto de Química, Universidad de Antioquia, Medellín, Colombia. santiago.mesa@udea.edu.co
b Instituto de Química, Universidad de Antioquia, Medellín, Colombia. johana.arboleda@udea.edu.co
b Instituto de Química, Universidad de Antioquia, Medellín, Colombia. adriana.echavarria@udea.edu.co
Received: July 17th, 2015. Received in revised form: February 24th, 2016. Accepted: March 7th, 2016
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.

Abstract:
In this paper zeolite Beta was synthesized and characterized by XRD, ATG, ATD, XRF and SEM. Subsequently various modifications were made using a surface passivation process in liquid phase with tetraethylorthosilicate (TEOS), using two loads of TEOS and three passivation cycles, in order to obtain six catalysts. These materials were characterized by NH3-TPD and tested in tolune disproportionation reaction, under different conditions of pressure and temperature. As modification cycles were increased for each catalyst, there was a change in the acidity of the solids, mainly due to deposition of an inert layer of silica on the external surface. These results were confirmed with the improvement in para-selectivity and decreasing in conversion. Despite zeolite Beta is a large pore zeolite, an improvement of the para-selectivity was obtained, achieving almost stable values for the entire range of temperature and pressure conditions.
Keywords: Beta zeolita, Surface Passivation, Catalysis, Toluene Disproportionation, Para-selectivity.
Resumen:
En este trabajo se sintetizó la zeolita Beta y se caracterizó por las técnicas de DRX, ATG, ATD, FRX y MEB. Posteriormente se realizaron diversas modificaciones mediante un proceso de pasivación superficial en fase líquida, usando tetraetilortosilicato (TEOS), utilizando dos cargas de TEOS y tres ciclos de pasivación, dando lugar a 6 catalizadores. Dichos materiales fueron caracterizados por medio de NH3-DTP y probados en la reacción de desproporción de tolueno, bajo diferentes condiciones de presión y temperatura. A medida que se incrementaron los ciclos de modificación para cada catalizador, se generó un cambio en la acidez de los sólidos debido principalmente a la deposición de una capa inerte de sílica sobre la superficie externa, corroborado con el mejoramiento en la para-selectividad y la disminución de la conversión de los sólidos modificados. A pesar que la zeolita Beta es una zeolita de poro grande, se logró un mejoramiento de la para-selectividad, logrando valores casi estables para todo el rango de condiciones evaluadas.
Palabras clave: Zeolita Beta, Pasivación Superficial, Catálisis, Desproporción de Tolueno, Para-selectividad
1. Introducción
Desde la década de 1950, la investigación en síntesis de zeolitas ha crecido significativamente, debido a sus amplias propiedades y al sinnúmero de aplicaciones que poseen estos materiales [1], dentro de las que se destacan los procesos de craqueo catalítico [2] y esterificación [3]. Dentro de sus propiedades se destaca la selectividad de forma. Esta característica permite la diferenciación, de acuerdo al tamaño, entre reactivos, productos e intermediarios de una reacción. Las moléculas que tengan las dimensiones adecuadas para entrar a los poros de las zeolitas, pueden ingresar y reaccionar allí. Por otro lado, las moléculas que puedan salir de los poros de las zeolitas, aparecerán como los productos finales. En este sentido, hay tres tipos de selectividad de forma [4]: i) Selectividad hacia reactivos: algunas moléculas que hacen parte de los reactivos iniciales son muy grandes y no se pueden difundir a través de los poros; ii) Selectividad hacia productos: algunos de los productos formados dentro de los poros de la zeolita son muy grandes y no se pueden difundir fuera de ellos; iii) Selectividad hacia el estado de transición: algunas reacciones no ocurren debido a que su correspondiente estado de transición requiere más espacio del que hay disponible dentro de las cavidades y poros de la zeolita. Las reacciones que pueden ocurrir sin impedimento son aquellas que requieren estados de transición pequeños.
A pesar de que una de las características principales de las zeolitas es su sistema de canales (por lo que posee una alta área superficial) y la mencionada selectividad de forma, la superficie externa está disponible para que cualquier molécula, independientemente de su tamaño, pueda reaccionar allí. Esto da lugar a un comportamiento catalítico no selectivo, por lo que el interés se ha volcado hacia la pasivación de los posibles sitios activos de la superficie externa.
La pasivación superficial externa de las zeolitas se ha investigado desde hace aproximadamente dos décadas, y generalmente se han usado técnicas en fase líquida, gaseosa e incluso reacción en fase sólida, usando habitualmente alcóxidos de silicio [5-9]. No solamente se ha apuntado hacia la pasivación superficial, sino también al mejoramiento de las propiedades catalíticas mediante el uso de algunos compuestos que puedan reaccionar sobre los sitios ácidos externos y generar capas activas. H. L. Janardhan y colaboradores [10] usaron la técnica de impregnación húmeda, y usando NH4H2PO4 lograron la deposición de una capa activa de fosfato, generando un nuevo tipo de sitios ácidos sobre la superficie del material. Dicho proceso de modificación ha tenido una amplia aplicación en reacciones que requieren selectividades considerables hacia cierto producto, por ejemplo la reacción de desproporción de tolueno selectiva [11]. Esta reacción es catalizada por zeolitas ácidas y usa tolueno como materia prima para transformarlo en benceno y xilenos [12]. La desactivación superficial de los sitios activos de la zeolita conduce a la formación selectiva de para-xileno dentro de la mezcla de xilenos formada en la reacción, y como bien es sabido, el isómero "para" es el más apetecido dentro de la mezcla de isómeros [13].
En este trabajo se sintetizó y caracterizó la zeolita Beta. Adicionalmente se le aplicó un proceso post-síntesis de pasivación superficial en fase líquida usando tetraetilortosilicato (TEOS). Se estudió la influencia del número de ciclos de modificación y la cantidad de TEOS sobre las propiedades catalíticas de los materiales en la reacción de desproporción de tolueno.
2. Metodología
2.1. Síntesis de la zeolita Beta
La síntesis de la zeolita Beta se realizó usando la composición molar Al2O3:947.2H2O:2Na2O:1K2O:52.9SiO2:26.2TEAOH. Inicialmente se prepararon dos soluciones, la primera (Solución 1) mezclando Hidróxido de Tetraetilamonio (35% w/w en H2O, Alfa Aesar), NaCl (99.8%, Sigma-Aldrich), KCl (99.5%, Sigma-Aldrich) y Ludox (30% w/w en H2O, Aldrich); mientras que la segunda (Solución 2) se preparó mezclando el agua, NaOH (98%, Sigma-Aldrich) y aluminato de sodio (grado técnico, Sigma-Aldrich). Posteriormente la solución 2 se agregó lentamente a la solución 1, para dar lugar a la formación de un gel altamente viscoso. Dicho gel se dejó en agitación durante una hora a temperatura ambiente. Finalmente el gel se dispuso en reactores que fueron llevados a la estufa a 135ºC durante 48 horas.
Con el fin de remover el agente estructurante, el material obtenido fue calcinado a 600ºC (con una rampa de 5ºC/min) durante 2 horas. Luego para la obtención de la zeolita Beta ácida, se adicionó 1 g de sólido en 50 ml de solución 0.2 M de NH4NO3 (Merck, 99%). La suspensión formada se dejó en agitación durante 3 horas a una temperatura entre 55 y 60ºC. Pasado este tiempo, el sólido se recuperó por filtración. Dicho proceso de intercambio se repitió 3 veces. Finalmente el material fue calcinado a 600oC durante 6 horas para dar lugar a la zeolita Beta ácida (H-Beta).
2.2. Pasivación superficial
La modificación de la zeolita H-Beta se realizó siguiendo el proceso reportado por Shourong Zheng [14], modificando algunos parámetros. El proceso se describe a continuación: el material en polvo se dispuso en un balón de fondo redondo y se le agregó la cantidad de tetraetilortosilicato (TEOS, 98%, Merck) correspondiente, además de ciclohexano (99.5%, Sigma-Aldrich) con una relación de 20 ml/gramo de sólido. La mezcla se sometió a calentamiento en un baño de aceite, bajo condiciones de reflujo (70-80ºC) durante 3 horas. Pasado este tiempo, se removió el reflujo y el sólido se recuperó por evaporación del solvente. Finalmente, el material se sometió a calcinación a 550ºC, con una rampa de 5ºC/min durante 2 horas para remover completamente el ciclohexano y dar lugar a la deposición de una capa inerte de sílica. Este proceso se repitió dependiendo del número de ciclos de modificación deseados.
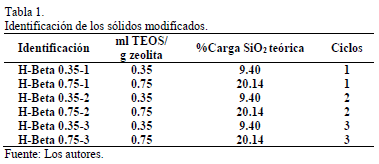
En la Tabla 1 se describen las condiciones usadas para la modificación de los sólidos, al igual que el nombre asignados para la identificación de cada uno de ellos. Los materiales fueron nombrados teniendo en cuenta la carga de TEOS usada y el número de ciclos a los que fueron sometidos. Por ejemplo, la zeolita H-Beta que se trató con 0.35 ml TEOS/g de sólido y fue sometida a un solo ciclo de pasivación fue nombrada H-Beta 0.35-1.
2.3. Caracterización
El análisis de difracción de rayos X (DRX) se llevó a cabo a temperatura ambiente en un difractómetro PANalytical Empyrean, usando una fuente de radiación de cobre (longitud de onda l=1,5418 Å), con un goniómetro Omega/2Theta, en un rango 2q de 5-50, a un paso de 0.02º en un modo de escaneo continuo.
La técnica de espectroscopia de absorción atómica se usó para el seguimiento del proceso de intercambio iónico en un espectrómetro Thermo Scientific iCE Series 3000.
Además, se realizaron análisis de fluorescencia de rayos X (FRX) para determinar la composición de los materiales sintetizados, usando un equipo Axios MAX (PANalytical), de longitud de onda dispersiva con fuente de Rodio.
El análisis térmico diferencial (ATD) se realizó en un equipo TA Instruments DSC 2920, en atmosfera de aire a 30 ml/min y un rango de barrido de 30 °C a 800 °C a una velocidad de calentamiento de 10 °C/min. El análisis termogravimetrico (ATG) se llevó a cabo con un termoanalizador TA Instruments Hi-Res TGA 2950 en atmosfera de aire a 40 ml/min, en un intervalo de temperatura de 30- 800°C y a una velocidad de 10 °C/min.
Las mediciones de adsorción de nitrógeno se llevaron a cabo en un equipo ASAP 2020 (Micromeritics) a una temperatura de nitrógeno líquido de -196.69ºC y a presiones relativas entre 0.01 y 0.99.
La acidez de los sólidos fue medida mediante la técnica de desorción de amoníaco a temperatura programada (NH3-DTP). Para ello se usó un reactor de cuarzo en forma de U, en el cual se dispone el catalizador, ubicado en un horno calentado por resistencias programables. El registro de las señales de adsorción y desorción de amoníaco se realizó mediante un detector de conductividad térmica dispuesto a 80ºC. Cada muestra fue pretratada a 150ºC bajo flujo de nitrógeno con el fin de eliminar agua e impurezas. Posteriormente la adsorción de amoníaco se llevó a cabo a 100ºC, hasta una completa saturación de la superficie del sólido. Finalmente la desorción se completó desde 40ºC hasta 500ºC a una velocidad de calentamiento de 10º/min, período en el cual se registraron las señales del detector.
Las medidas de 27Al RMN se realizaron en un equipo Bruker FOURIER de 300 MHz, referenciando los desplazamientos relativos al nitrato de aluminio.
2.4. Ensayos catalíticos
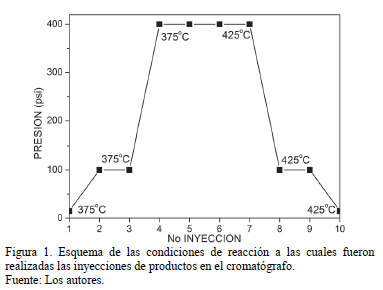
Para la reacción se utilizó hidrógeno como gas de arrastre, y tolueno líquido. El tolueno y el hidrógeno son conducidos hacia un reactor tubular de acero inoxidable el cual es calentado por un horno cilíndrico con control de temperatura. Allí se ponen en contacto con un gramo de catalizador con tamaño de partícula entre 425 y 600 mm. Los productos derivados de la reacción son conducidos hacia el cromatógrafo por una línea precalentada para evitar condensación. La reacción fue trabajada a 375 y 425ºC y desde presión atmosférica hasta 400 psi, con flujo de hidrógeno de 50 ml/min y de 0.1 ml/min de tolueno; dando lugar a una velocidad espacial (WHSV) de 5.2 h-1.
Con el fin de conocer el comportamiento catalítico de la reacción se estableció la siguiente metodología: como primera instancia, el catalizador se dejó activando una hora dentro del reactor bajo atmósfera de hidrógeno a 80 ml/min y a 375ºC. Luego de pasado este tiempo, se comenzó a inyectar tolueno a un flujo de 0.1 ml/min e hidrógeno a 50 ml/min; dejando estabilizar el sistema durante 30 minutos. Después de este lapso de tiempo, se realizó la primera inyección de los productos de reacción hacia el cromatógrafo. Con el fin de conocer el comportamiento catalítico de los materiales a diferentes condiciones se realizó un barrido de presión y temperatura de reacción, partiendo desde presión atmosférica hasta 400 psi y desde 375 hasta 425ºC, realizando dos inyecciones cromatograficas por cada condición de presión y temperatura establecida. De esta forma se probaron 3 presiones (14.7, 100 y 400 psi) por cada temperatura (375 y 425ºC), de acuerdo a lo que se presenta en la Fig 1.
Los productos de reacción fueron analizados en un cromatógrafo Agilent 7820. Los compuestos de la reacción fueron separados usando una columna HP-PONA, determinados usando un detector FID y usando flujos de 400 ml/min de aire, 40 ml/min de hidrógeno y 30 ml/min de nitrógeno.
Los porcentajes de conversión (%X), rendimiento (%R) y para-selectividad (%p-S) fueron calculados usando las siguientes ecuaciones:

3. Resultados y discusión
3.1. Síntesis y Caracterización
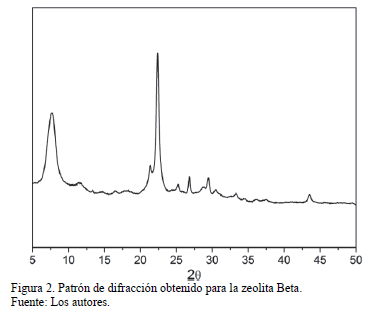
Según los resultados de difracción de rayos X (Fig 2), se pudo establecer que la zeolita Beta sintetizada efectivamente corresponde con el patrón estándar reportado para dicho sólido [15]. La estructura de la zeolita Beta posee una serie de señales a 7.6, 13.4, 14.4, 21.2 y 22.2 [16], que pudieron ser observadas en el patrón de difracción de la zeolita obtenida.
De acuerdo con los resultados de fluorescencia de rayos X, mostrados en la Tabla 2, se observa que efectivamente hubo una incorporación del silicio y el aluminio en la zeolita sintetizada. Sin embargo cabe resaltar que el rendimiento de la reacción de síntesis fue relativamente bajo, dado que la relación Si/Al experimental es menor que la calculada teóricamente a partir de la relación molar del gel, y por ende el silicio y el aluminio no se incorporaron totalmente a la red zeolítica.

La relación entre los parámetros de síntesis, las características estructurales y rendimientos del proceso de zeolitización no está del todo clara. Dicho proceso es térmicamente activado y usualmente se da a elevadas temperaturas, para producir cristales en un período de tiempo aceptable. Sin embargo, al parecer la nucleación está controlada más por aspectos cinéticos que por los termodinámicos [17]. Esto conduce a resaltar que los mecanismos de formación de zeolitas son complejos debido a la conjugación de reacciones, equilibrios y variaciones en las solubilidades que ocurren en la mezcla de reacción [1]. Adicionalmente, en Verified Synthesis of Zeolitic Materials [17] se pueden encontrar valores elevados de rendimientos, como ocurre para la zeolita Y, con un 98% basado en Al2O3; en contraste con otros rendimientos más bajos como el de la ZSM-35, perteneciente a la familia FER, que alcanza un 60% basado en SiO2.
En la microscopía electrónica de barrido (Fig 3) se puede observar que la zeolita Beta obtenida presenta una morfología esférica, con formaciones de grandes conglomerados, la cual es una morfología comúnmente observada para la zeolita Beta [17,18]. Dichas agrupaciones no presentan fases crecientes parásitas o contaminaciones, según la micrografía. El tamaño de partícula aproximado es de 540 nm.

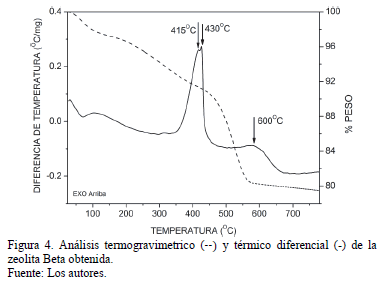
Los análisis térmicos del la zeolita Beta obtenida se presentan en las Fig 4. En el análisis termogravimétrico se observó un primer evento térmico debido a la pérdida de humedad y agua ocluída en los poros de la zeolita. La segunda pérdida se puede dividir en dos etapas: a baja temperatura (250-400ºC), la cual es el resultado de la evolución de los cationes TEA+ (dando lugar a trietilamonio y etileno) que no están asociadas a la estructura zeolítica como tal y que pueden hacer parte de los defectos estructurales de la red [19]. La otra etapa (a alta temperatura, >400ºC) se le asigna la pérdida de los cationes TEA+ que balancean las especies Al(SiO)4- de la red, ya que dichos cationes están más fuertemente enlazados a la estructura [19]. Ambos eventos exotérmicos son diferenciables en el ATD (Entre 415-430ºC y a 600ºC aproximadamente) presentado en la Fig 5. La pérdida total en peso alcanzó un valor de 20.5%.
3.2. Zeolita Beta ácida (H-Beta) y Caracterización
Una vez el sólido fue sintetizado y calcinado, se sometió al proceso de intercambio iónico para dar lugar a su forma ácida (H-Beta). En la Tabla 3 se presentan los resultados para sodio y potasio antes y después del intercambio, obtenidos mediante absorción atómica.

Se puede observar que el porcentaje de intercambio alcanzado fue mayor al 90%, llegando a un valor cercano al 100% para el caso del potasio. Ello da cuenta de un proceso de intercambio exitoso, que combinado con la etapa de calcinación posterior dio lugar a una activación ácida casi completa del sólido. Adicionalmente, el patrón de difracción de rayos X de la zeolita Beta ácida no presentó cambios significativos en comparación con el sólido sintetizado.
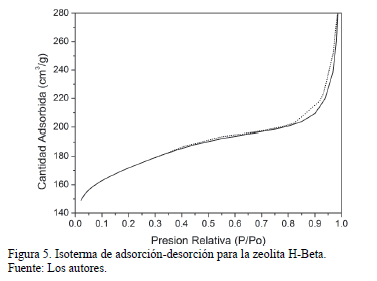
El análisis de adsorción de nitrógeno de la zeolita beta ácida se muestra en la Fig 5, presentando una isoterma de adsorción del tipo I según la clasificación de la IUPAC [20]. Las sustancias microporosas con tamaños de poros por debajo de los 20 Å exhiben este tipo de isotermas. La curva de adsorción pronunciada que se presenta a bajas presiones es debido a la condensación capilar del nitrógeno en los microporos del material. Después del llenado de los microporos, la adsorción solo puede ocurrir en la superficie externa y por ende la curva se estabiliza [21]. El área superficial del material fue de 629.36 m2/g, con un área externa 193.85 m2/g, lo cual está en concordancia con lo encontrado en la literatura [18,22].
3.3. Sólidos pasivados y caracterización
Los análisis químicos por fluorescencia de rayos X (FRX) de los sólidos pasivados se presentan en la Tabla 4. Se observa que en general ocurrió una disminución constante en la cantidad de aluminio a medida que se incrementa el número de ciclos y la carga de TEOS, dando como resultado un incremento en la relación Si/Al obtenida por FRX para los materiales modificados, en comparación con la zeolita sin modificar. Adicionalmente se depositaron pequeñas cantidades de silicio sobre los sólidos, de tal forma que el material con mayor modificación (H-Beta 0.75-3) tuvo el mayor incremento en la cantidad de SiO2 con respecto al material sintetizado. Por otro lado se presentó una particularidad para los materiales H-Beta 0.75-1, H-Beta 0.35-2 y H-Beta 0.75-2: a medida que se incrementó la cantidad de TEOS, disminuyó el porcentaje de silicio en los sólidos (especialmente para los sólidos tratados con 2 ciclos). En este sentido, se cree que no hay lugar a una desilicación de la zeolita tratada, debido a que para ello se necesitan pH altamente básicos y un tratamiento diferente al que se plantea en este trabajo [23]. Es por ello que se infiere que la disminución en los porcentajes de silicio puede deberse a la relación en peso de los sólidos pasivados en comparación con el sólido ácido.
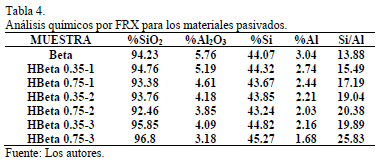
Igualmente, para los pequeños incrementos en la cantidad de sílica depositada (en comparación con la carga teórica), podría explicarse de dos maneras: 1) el agua residual presente en el TEOS o en la misma zeolita generó una hidrólisis prematura de la molécula orgánica [6], y 2) reacción del TEOS con el aluminio extra-estructural, resultando en una baja adsorción de la molécula orgánica sobre la superficie de la zeolita. Al respecto, Hui Teng [11] sugiere un tratamiento ácido antes del proceso de pasivación. Los ácidos orgánicos poseen un tamaño mayor que el poro de la zeolita, y pueden ser usados para remover selectivamente aluminio extra-estructural, dando lugar a una superficie rica en silicio, lo que puede beneficiar el proceso de deposición de la capa inerte de sílica [11].
En la Fig. 6 se muestran los análisis de desorción de amoníaco a temperatura programada de los catalizadores probados. De manera general, se observa que todos los materiales analizados presentan dos picos de desorción, uno a baja temperatura (150-250ºC) y otro a alta temperatura (350-450ºC), correspondientes a los sitios ácidos débiles y fuertes, respectivamente [24]. Los catalizadores modificados presentaron menor acidez que la zeolita ácida sin modificar. Aunque el efecto no es muy notorio para un solo ciclo de modificación, se observa que el primer pico de desorción para el material H-Beta 0.35-1 ocurre a una temperatura un poco menor (178ºC aproximadamente) que el del material sin modificar (210ºC); esto daría cuenta de una menor fuerza ácida del material modificado.
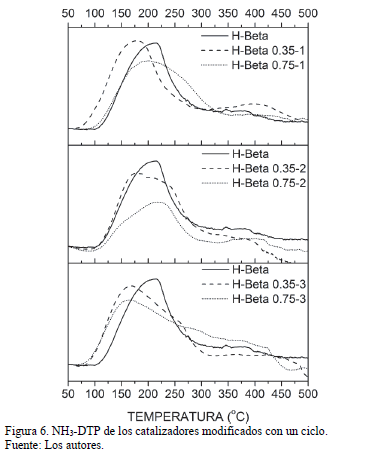
Adicionalmente, el pico de desorción de alta temperatura del catalizador H-Beta 0.35-1 es mayor que el del catalizador sin modificar, ello podría deberse al fenómeno conocido "Next-Nearest Neighbours", en el cual se expone que mientras más alejados entre sí estén los sitios ácidos, mayor fuerza ácida van a presentar, aunque la acidez total del sólido se ve disminuida [25].
Por otro lado, el pico de desorción correspondiente a los sitios ácidos débiles del catalizador H-Beta 0.75-1 es más amplio que el del sólido sin modificar, indicando una generación de sitios ácidos débiles debido posiblemente a la dealuminización causada por el proceso de modificación.
El catalizador H-Beta 0.35-2 presenta menor acidez que el sólido sin modificar, confirmando que efectivamente la pasivación genera una desactivación de los sitios ácidos. El efecto más notorio se da en los sitios ácidos débiles, siendo el catalizador H-Beta 0.75-2 el que presenta mayor disminución. No es posible afirmar algo concluyente sobre los sitios ácidos fuertes dado que la línea base de algunos catalizadores probados fueron inestables y oscilantes.
Un efecto interesante se observó con los sólidos pasivados con tres ciclos de modificación: a medida que se incrementó la carga de TEOS, el pico de desorción asociado a los sitios ácidos débiles se vuelve más ancho, indicando que las moléculas de amoníaco son desorbidas más lentamente. Este fenómeno tiene explicación en base a la dealuminización causada por el proceso de modificación, lo que resultó en una deposición de aluminio extra-estructural, y una generación de mayor densidad de sitios ácidos débiles [24].
El catalizador menos modificado (H-Beta 0.35-1) presentó una acidez total de 764.1 mmol/g, mientras que el más modificado (H-Beta 0.75-3) obtuvo un valor de 459.9 mmol/g; presentándose una disminución de la acidez de 39%, lo que indica que los ciclos de modificación afectan fuertemente el grado de pasivación superficial. Sin embargo no fue posible garantizar la pasivación superficial sin que ocurriera dealuminización. Los análisis de 27Al RMN que se realizaron a los catalizadores H-Beta 0.35-1 y H-Beta 0.75-3 (Figuras no presentadas), mostraron dos bandas: una a 50 ppm correspondiente a los aluminios tetraedrales que hacen parte de la red zeolítica, mientras que la banda en aproximadamente -10 ppm está ligada al aluminio octaédrico, correspondiente al aluminio generado por dealuminización [18]. La relación de aluminios tetraédricos/octaédricos para el material H-Beta 0.35-1 es de 2.5/1; mientras que la relación para el sólido H-Beta 0.75-3 es de 2/1, lo que indica que efectivamente hubo una dealuminización a medida que se incrementaron los ciclos de modificación.
3.4. Catálisis
Inicialmente, en la Fig. 7 se observa que el catalizador sólido no pasivado H-Beta presenta grandes valores de conversión en todo el rango de temperatura y presión evaluadas. Los poros grandes y el carácter tridimensional de los canales de la zeolita Beta hacen que sea muy activa en la reacción, lo que genera que el tolueno llegue más fácilmente al interior del sólido, disminuyendo las limitaciones difusionales, y logrando valores de conversión de tolueno cercanos al 60% a 400 psi y 425ºC. Sin embargo, es de notar que los catalizadores modificados con un ciclo (H-Beta 0.35-1 y H-Beta 0-75-1), tuvieron conversiones mayores que el sólido sin pasivar, lo que confirma el comportamiento observado en los análisis de acidez, donde se observó una generación de sitios ácidos débiles, que posiblemente promovieron la conversión de tolueno. En este sentido, por medio del análisis de desorción de amoníaco no es posible confirmar si dichos sitios ácidos débiles son sitios ácidos del tipo Brønsted o Lewis, sin embargo se sabe que la reacción de desproporción de tolueno es catalizada por sitios ácidos del tipo Brønsted.
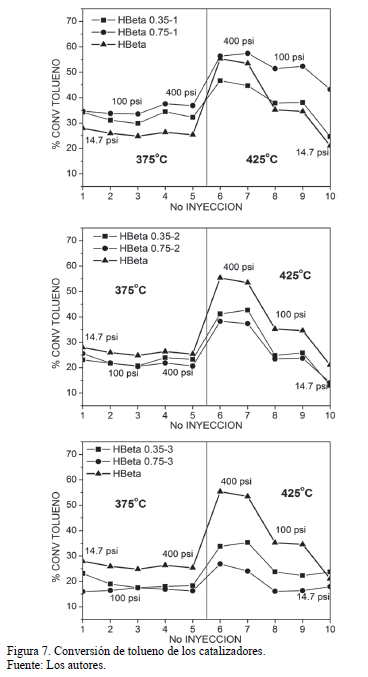
Por otra parte, dos y tres ciclos de modificación fueron suficientes para lograr una disminución significativa de la conversión, logrando que se depositara una capa inerte de sílica. De esta forma, el sólido sometido a más ciclos de pasivación y más carga de TEOS (H-Beta 0.75-3) fue el que obtuvo la menor conversión de tolueno.
Como era de esperarse el incremento en la presión y la temperatura generaron mayor conversión de tolueno por parte de los catalizadores. Un aumento en la presión ocasiona que el tolueno sea forzado a ingresar a los poros de la zeolita y por ende se mejore la adsorción sobre los sitios activos del catalizador [26]. En este sentido se puede observar que el incremento en la presión genera pequeños aumentos en la conversión a 375ºC, mientras que un incremento en la temperatura (de 375 a 425ºC) genera grandes cambios en la conversión, debido al carácter endotérmico de la reacción.
Así mismo, no se observó una desactivación de los sólidos por coquización, debido a la presencia de hidrógeno como gas de arrastre [26]. Los catalizadores siguieron presentando actividad a condiciones de 425ºC y presión atmosférica (condiciones de reacción de la última inyección de productos).
En la Fig 8 se muestran los rendimientos hacia xilenos de cada uno de los catalizadores ácidos. La zeolita H-Beta presentó un rendimiento de aproximadamente 25% a condiciones de 400 psi y 425ºC. También se observó que los sólidos modificados presentaron menores valores de rendimiento, siendo el catalizador con más ciclos de pasivación (H-Beta 0.75-3) con el menor rendimiento, confirmando la reducción en su actividad catalítica por cuenta de los procesos de pasivación. Sin embargo, es de notar que los valores de rendimiento de los sólidos pasivados con un solo ciclo, no difieren considerablemente del sólido ácido sin modificar, lo que sugiere la alta actividad que sigue presentando el sólido después de solo un ciclo de pasivación. A este respecto, Paul J. Kunkeler y colaboradores [8] indican que la superficie externa de la zeolita Beta puede ser extremadamente activa, y por ende es necesario la aplicación de procesos de modificación post-síntesis para reducir la actividad superficial, con el fin de obtener materiales catalíticos con la acidez apropiada. En este sentido, las disminuciones más fuertes en los valores de rendimiento se presentaron para los catalizadores modificados con dos y tres ciclos, bien sea usando cargas de 0.35 ó 0.75 ml/g de zeolita.
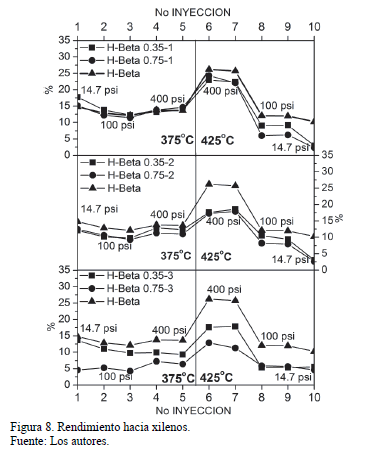
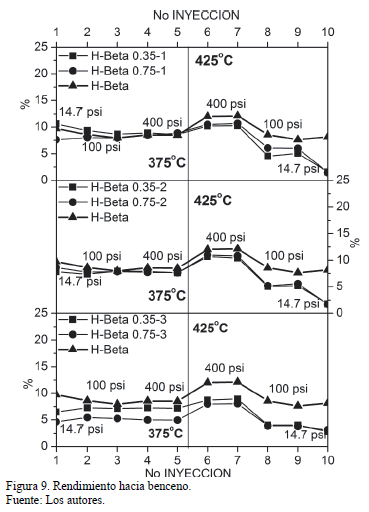
El rendimiento hacia benceno se presenta en la Fig. 9. Se observa una misma tendencia que el rendimiento hacia xilenos: el efecto más pronunciado sobre dicho rendimiento se da por cuenta de la temperatura. Sin embargo, el incremento en la presión a 375ºC generó una ligera disminución en el rendimiento del catalizador sin modificación. Al respecto, S. Al Khataff y colaboradores [26] afirman que a pesar que el incremento en la presión genera un aumento en la cantidad de benceno debido a las reacciones laterales, la presencia de hidrógeno tiende a prevenir dichas reacciones, e incluso llegando a obtenerse valores menores de selectividad y rendimiento hacia benceno al incrementar la presión.
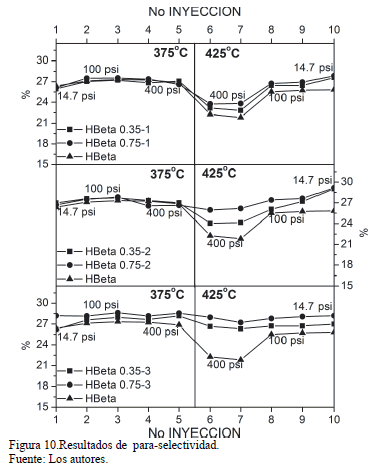
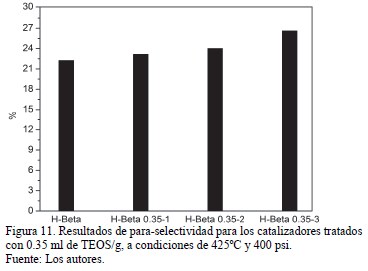
Las selectividades hacia para-xileno en la mezcla de xilenos se muestran en la Fig 10. La para-selectividad de los catalizadores aumentó a medida que se aumentó el número de ciclos de modificación como se observa en la Fig 11. Al incrementar la presión (de 100 a 400 psi) a 375ºC no hay un cambio significativo en los valores de para-selectividad, y el efecto de la presión sobre dichos valores es más notorio a 425ºC. Sin embargo, al incrementar la temperatura de 375 a 425ºC a 400 psi, hay una disminución sustancial en los valores de para-selectividad, debido al incremento de productos derivados de reacciones laterales [26]. No obstante, el efecto de la temperatura es menos notorio a medida que se incrementan los ciclos de modificación, sin importar la carga de TEOS usada (0.35 ó 0.75 ml/g), logrando valores casi estables para el catalizador H-Beta 0.75-3. Esto podría atribuirse no solo a la pasivación de los sitios activos externos, sino también a un estrechamiento de la entrada del canal por acción de las moléculas de TEOS. El diámetro cinético del TEOS es mayor que la entrada de los poros de la zeolita, y algunos grupos hidroxilo que están cerca a la entrada de los canales pueden reaccionar, para dar lugar a enlaces Si-O-Al ó Si-O-Si [11].
Además, la zeolita Beta es clasificada como una zeolita de poro grande, y para este caso, se depositaría una capa de sílica inerte en la entrada de los canales lo que generaría un estrechamiento, dando lugar a que los productos intermediarios "grandes" no puedan salir de los canales. De hecho el rendimiento hacia productos C9+ a condiciones de 425ºC y 400 psi disminuyó desde 15% para el catalizador H-Beta, hasta un 3.5% para el catalizador H-Beta 0.75-3.
Con el fin de de medir la cantidad de coque depositada en los catalizadores luego de la reacción, los sólidos H-Beta y H-Beta 0.75-3 fueron sometidos a un programa de análisis termogravimétrico. Los resultados se observan en la Fig 12. Fue evidente que el sólido modificado presentó menor cantidad de coque depositado luego de la reacción, en comparación con el sólido sin pasivación.
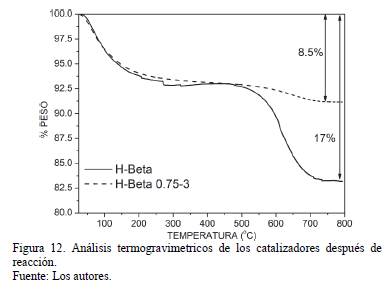
Este hecho, combinado con el mejoramiento de la para-selectividad, indica que el sólido pasivado inhibió la formación de compuestos grandes, como etiltolueno y trimetilbenceno, que pueden formarse fuera de los poros del catalizador para dar lugar a la formación de coque.
4. Conclusiones
El método hidrotérmico es un método viable para la obtención de la zeolita Beta; esto se corroboró mediante difracción de rayos X, donde se pudo evidenciar que el difractograma obtenido experimentalmente era congruente con su homólogo simulado. Igualmente se pudo corroborar mediante absorción atómica que el intercambio iónico combinado con la etapa de calcinación posterior dio lugar a una activación ácida casi completa del sólido.
Por otro lado, fue posible realizar modificaciones a la zeolita Beta ácida, mediante un método de pasivación superficial, usando varias cargas de TEOS y varios ciclos de modificación, dando lugar a materiales con diferentes características fisicoquímicas. Precisamente, se evidenció mediante 27Al RMN que las modificaciones generaron una pequeña dealuminización debido posiblemente a las condiciones de modificación. Sin embargo, el cambio en la acidez de los sólidos es debido principalmente a la deposición de una capa inerte de sílica sobre la superficie externa, corroborado con el mejoramiento en la para-selectividad de los sólidos modificados y la disminución en la cantidad de coque depositada sobre el catalizador H-Beta 0.75-3. Además, a medida que se incrementaban los ciclos de modificación para cada catalizador, se incrementó la para-selectividad (especialmente para dos y tres ciclos) a la vez que se disminuía la conversión de tolueno, lo que indica que efectivamente los procesos de modificación lograron el bloqueo de los sitios activos externos del catalizador.
A este respecto es de notar que la zeolita Beta es una zeolita de poro grande, y por ende lograr conversiones selectivas hacia para-xileno se hace complicado. Sin embargo, mediante el proceso de pasivación se logró un mejoramiento de la para-selectividad, logrando valores casi estables para todo el rango de condiciones evaluadas.
Un ciclo de modificación no fue suficiente para lograr una pasivación notable de los sólidos. Incluso se notó que se generó mayor fuerza ácida de lo sólidos, gracias al efecto conocido como "Next-Nearest Neighbours". Por cuenta de ello, los valores de conversión de tolueno para los sólidos modificados con solo un ciclo de modificación, fueron ligeramente mayores que para el sólido sin modificación. Además, los incrementos de temperatura y presión de la reacción, generó aumentos en la conversión de tolueno. Dicho fenómeno se presenta por el incremento de reacciones laterales y por el carácter endotérmico de la reacción.
Agradecimientos
Los autores agradecen el apoyo financiero recibido por parte del Instituto Colombiano del Petróleo y al Programa de Sostenibilidad 2014-2015 de la Universidad de Antioquia.
Referencias
[1] Byrappa, K. and Yoshimura, M., Handbook of Hydrothermal Technology. New York. William Andrew Publishing/Noyes, 2001. DOI: 0.1016/B978-081551445-9.50001-5. [ Links ]
[2] Nancy, T.R. and Janneth. T.A., Using spent fluid catalytic cracking (FCC) catalyst as pozzolanic addition - A review. Ingeniería e Investigación, 30(2), pp. 35-42, 2010. [ Links ]
[3] Sanchez F.J. and Niño C.J., Influence of the aluminium impregnation [Al(NO3)3] in the beta zeolite over its acidity. Ingeniería e Investigación, 24(2). pp. 51-61, 2010. [ Links ]
[4] Ernst, S., Advances in nanoporous materials. 1, Elsevier, 2009. DOI: 10.1016/S1878-7959(09)00107-8. [ Links ]
[5] O'Connor, C.T., Moller, K.P. and Manstein, H., The effect of silanisation on the catalytic and sorption properties of zeolites. KONA, 25, pp. 230-236. 2007. DOI: 10.14356/kona.2007020. [ Links ]
[6] Weber, R.W., Moller, K.P. and O'Connor, C.T., The chemical vapour and liquid deposition of tetraethoxysilane on ZSM-5, mordenite and beta. Microporous and Mesoporous Materials, 35-36, pp. 533-543. 2000. DOI: 10.1016/S1387-1811(99)00248-6. [ Links ]
[7] Ren, X., Liang, J. and Wang, J., H-MCM-22 zeolitic catalysts modified by chemical liquid deposition for shape-selective disproportionation of toluene. Journal of Porous Materials, 13, pp. 353-357, 2006. DOI: 10.1007/s10934-006-8030-5%I [ Links ]
[8] Kunkeler, P.J., Moeskops, D. and van Bekkum, H., Zeolite Beta: Characterization and passivation of the external surface acidity. Microporous Materials, 11(5-6), pp. 313-323. 1997. DOI: 10.1016/S0927-6513(97)00053-9. [ Links ]
[9] Zheng S., Jentys, A. and Lercher, J.A., On the enhanced para-selectivity of HZSM-5 modified by antimony oxide. Journal of Catalysis, 219, pp. 310-319, 2003. DOI: 10.1016/S0021-9517(03)00234-3. [ Links ]
[10] Janardhan, H.L., Shanbhag, G.V. and Halgeri, A.B., Shape-selective catalysis by phosphate modified ZSM-5: Generationof new acid sites with pore narrowing. Applied Catalysys A: General, 471, pp. 12-18, 2014. DOI: 10.1016/j.apcata.2013.11.029. [ Links ]
[11] Teng H., Wang J., Ren X. and Chen D., Disproportionation of toluene by modified ZSM-5 zeolites catalysts with high shape-selectivity prepared using chemical liquid deposition with tetraethyl orthosilicate. Chinese Journal of Chemical Engineering, 19(2), pp. 292-298, 2011. DOI: 10.1016/S1004-9541(11)60168-7. [ Links ]
[12] Tsai, T-Ch., Liu, S-B. and Wang, I., Disproportionation and transalkylation of alkylbenzenes over zeolite catalysts. Applied Catalysis A: General, 181(2), pp. 355-398, 1999. DOI: 10.1016/S0926-860X(98)00396-2. [ Links ]
[13] Wiley, Petroleum Technology. Vol. 2.John Wiley & Sons. New York. 2007. [ Links ]
[14] Zheng, S., Surface modification of HZSM-5 zeolites. Tesis de Dr. der Naturwissenschaften. Institut fur Teschnische Chemie Il der Teschnischen Universitat Munchen. Munich, Germany, 2002. [ Links ]
[15] Wei, H., Liu, K., Xie, S., Xin,W., Li, X., Liu, S. and Xu, L., Determination of different acid sites in Beta zeolite for anisole acylation with acetic anhydride. Journal of Catalysis, 307, pp. 103-110, 2014. DOI: 10.1016/j.jcat.2013.07.010. [ Links ]
[16] Talebi, G., Amrollahi, M. and Fathizadeh, M., Adsorption of benzene from benzene/n-C6 and n-C7 mixture by nano Beta zeolite with different Si/Al ratios. Arabian Journal of Chemistry, In-Press. 2014. DOI: 10.1016/j.arabjc.2014.08.015. [ Links ]
[17] Robson, H. and Lillerud, K.P., Verified Synthesis of Zeolitic Materials. Elsevier. New York. 2001. [ Links ]
[18] Zarama-Pazos, M.C., Contribución a la sintesis hidrotérmica de zeolita Beta y sus posibles modificaciones con Galio. Tesis MSc.en Ciencias Químicas. Universidad Nacional de Colombia, Bogotá, Colombia, 2004. [ Links ]
[19] Clambor, M.A., Corma, A. and Valencia, S., Characterization of nanocrystalline zeolite Beta, Microporous and Mesoporous Materials, 25(1-3), pp. 59-74, 1998. DOI: 10.1016/S1387-1811(98)00172-3. [ Links ]
[20] Gregg, S.J. and Sing, K.S.W., Adsorption, surface area and porosity. Academic Press. Michigan. 1991. [ Links ]
[21] Abrevaya, H., Unique aspects of mechanisms and requirements for Zeolite catalysis in refining and petrochemical, In: Kulprathipanja, S. Catalysis Zeolites in Industrial Separation, Glasgow, John Wiley & Sons, 2010, pp. 406-581. [ Links ]
[22] Muller, M., Harvey, G. and Prins, R.,Quantitative multinuclear MAS NMR studies of zeolites. Microporous and Mesoporous materials, 34(3), pp. 281-290, 2000. DOI: 10.1016/S1387-1811(99)00180-8. [ Links ]
[23] Groen, J.C., Abelló, S., Villaescusa, L.A. and Pérez-Ramírez, J., Mesoporous beta zeolite obtained by desilication. Microporous and mesoporous materials, 114(1-3), pp. 93-102, 2008. DOI: 10.1016/j.micromeso.2007.12.025. [ Links ]
[24] Damjanovic, L. and Auroux, A., Determination of Acid/Base properties by temperature programmed desorption (TPD) and adsorption calorimetry, In: Derouane, E.G. and Chester, A.W. Eds. Zeolite Characterization and Catalysis: A Tutorial. Elsevier, pp. 107-167, 2009. DOI: 10.1007/978-1-4020-9678-5_3. [ Links ]
[25] Scherzer, J., Correlation between catalyst formulation and catalytic properties, In: Magee, J.S. and Mitchell, M.M., Eds. Fluid catalytic cracking: Science and Technology. Elsevier, pp. 145-182. 1993. DOI: 10.1016/S0167-2991(08)63828-8. [ Links ]
[26] Al-Khattaf, S., Ali, M.A. and Al-Amer, A., Effect of reaction pressure and carrier gas on toluene disproportionation over molybdenum-ZSM-5 catalyst. Energy & Fuels, 22(1), pp. 243-249, DOI: 10.1021/ef700582b. 2008. [ Links ]
S. Mesa, es Ingeniero de Materiales desde el 2011, recibió su grado de MSc. en Ciencias Químicas en el año 2015, ambos de la Universidad de Antioquia, Colombia. Desde el año 2012 hasta el año 2015 trabajó en investigación en catálisis usando mallas moleculares en procesos de la industria petroquímica. Sus áreas de investigación son: zeolitas y mallas moleculares, síntesis inorgánica, catálisis, petroquímica y energía. ORCID: 0000-0002-0058-7193
J. Arboleda, recibió grado de Ingeniera Química en 2006 y de Dra. en Ciencias Químicas en 2010, de la Universidad de Antioquia, Colombia. Se ha desempeñado como docente en el área de química analítica e intrumental y como investigadora en las áreas de caracterización de nuevos materiales, aplicaciones de nuevos materiales en procesos catalíticos y microscopía electrónica. Sus áreas de investigación son: síntesis inorgánica, catálisis, petroquímica y energía, microscopia electrónica. ORCID: 0000-0003-0588-6093
A.Echavarría-Isaza, obtuvo su grado de Ingeniera Química en el año 1993, su título de MSc. en Ciencias Quimicas en 1997 y el de Dra. en Ciencias Químicas en 2001, de la Universidad de Antioquia, Colombia. Se ha desempeñado como docente titular de la Universidad de Antioquia en las áreas de catálisis, química inorgánica y química analítica. Sus investigaciones han estado enfocadas en las áreas de zeolitas y su uso en la industria petroquímica, caracterización de materiales y catálisis heterogénea. ORCID: 0000-0002-1733-9583

