











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
1. Introduction
In recent years, new materials that exhibit electronic and optical properties that are modulated by various parameters have been developed. In this context, YxIn1-xN is a material that is potentially attractive for industry, because it is made up of two materials, InN and YN, which exhibit very different behavioral characteristics under external hydrostatic pressure: the optical gap of InN increases with increasing pressure, while that of YN decreases [1]. Thus, it is to be expected that for certain concentrations of yttrium, the optical gap would be constant upon varying the pressure. This behavior would be useful in devices that work under highly variable pressure. The nitride group III-V is based on the first transition metals, which possess physical properties of a high degree of electron mobility, high degree of hardness, low fragility, stability at high temperatures, high melting point, and good thermal and electrical conductivity, which implies a potential for use in the development of optoelectronic devices, devices for magnetic storage, hard coatings for cutting tools, and protection against corrosion [1,7,8,17].
In a previous study, Abdel Rahim Garzón, G. P. [1,8] found that in its ground state the crystallization and lattice parameters for YN is B1, with ~4.91 Å, and for InN it is B4, with ~3.58 Å. The bulk moduli are B 0, ~163 GPa and B 0 , ~119 GPa, respectively, and the volume increases as the compound has more d valence electrons and a greater presence of electrons in the inner layers, so we can reasonably assume that the natural crystallization phase for YxIn1-xN will be between these two phases. Abdel Rahim Garzón, G. P. and Jairo Arbey Rodríguez Martínez [8,9], using DFT, studied the structural and electronic properties of the compound YInN in the B2 and B1 phases, with concentrations x of ¼, ½, and – of Y. It was found that in the B1 phase, at a concentration of – yttrium (Y) the material behaves as a semiconductor, while in the B2 phase it is metallic at the three concentrations. The cohesion energy decreases with increasing Y concentration, and this is observed for both phases, and the gap energy depends on the yttrium concentration.
They found that for the compound Y0.5In0.5N, the ground state is B4, with a difference of energies between the minima of the structures B1 and B4 of ~0.025 eV/unit formula, with a possible B4 to B1 phase transition of the structure at a pressure of PT ~1.36 GPa. In the band structure, it was observed that at P=0 in the B1 phase there is a metallic behavior, and in the B4 phase and in the phase transition from the semiconductor to the metal phase an indirect semiconductor behavior is exhibited [8,10]. Additionally, they studied the compound YIn3N4 in the B1 and B4 phases and found that the base state of the crystallization for this compound is B4, with a possible phase transition of the structure from B4 to B1 with a pressure of PT ~5.6 GPa. The B1 phase exhibits metallic behavior, while the B4 phase is a semiconductor with a gap energy of ~0.2 eV [8,11].
Zoita et al. [13] reported on the synthesis of YxIn1-xN with low levels of Y (x<0.1), experimentally determining that the base state is WZ, and on increasing the melting point of the material, the lattice constant increases and the bandwidth increases linearly towards blue. Also, they determined that the base state of YN is the B1 phase, with an indirect energy gap of ~0.498eV. Tholander et al. [2] report that the composition of thin films with Y (content up to x=0.5) depends on the lattice parameters, and using ab initio calculations they determined that the YxIn1-xN wurtzite phase is the lowest in energy among the relevant alloy structures for 0<x≤0.5. Additionally, it was determined that this compound used as films on AlN not only reduces surface roughness but also improves the crystalline quality. Also, the piezoelectric properties of YInN were improved, converting it into a possible candidate for future electroacoustic applications. In addition, in a theoretical study by N. Benyounes et al. [5], YN was suggested as a possible alloy for tuning the direct and indirect bandgap in metastable zincblende (B3) InN.
In the present paper, the NaCl-type (B1), CsCl-type (B2), ZnS-type (B3), and WZ-type (B4) phases of YxIn1-xN are studied as first step for a later study in which it will be determined in which phase the compound is energetically more stable or metastable, and the potential changes of phase will be investigated. Also, a detailed study of its structural stability in the four phases is presented. Plots of energy vs. volume at T = 0K are shown, and some thermodynamic and structural properties are determined, such as the volume unit, B = -V(∂P/∂V) T, its derivative, the cohesive energy, and the unit cell volume. With respect to the electronic structure, a detailed analysis is made of the densities of state (DOS), and the band structure is plotted in order to determine the behavior of the direct and indirect bandgap. This article is organized as follows: First, the method that is used is described; in the following section, a study of the stability and the electronic properties is presented for YN, InN, and YxIn1-xN; and last, a summary is presented, along with some conclusions with respect to YxIn1-xN.
2. Computational methods
The calculations were performed using density functional theory (DFT), with the full potential linearized augmented plane wave (FP-LAPW) method, as implemented in wien2k code [14]. The exchange and correlation effects are dealt with by using the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof parametrization [15]. For all concentrations of x, the Y parameters are: Y: muffin-tin radius = 1.8 u.a., In; muffin-tin radius = 1.85 u.a, and N: muffin-tin radius = 1.6 u.a, in addition to other values used, such as: lmax= 10.0. lmax is the factor that determines the maximum value of l. R kmax = 8.0, R kmax determines the size for the base of φk functions and is equal to R kmax =R mt ×K max , where K max is the magnitude of the largest G vector, and R m is the muffin-tin radius of the smallest sphere. The energy of separation between the shell and the valence states is -8.0 Ry. Total number of 0.25 vectors in the first Brillouin zone is k = 1000 for all phases. Convergence criterion in the energy of the self-consistent cycles is 0.0001 Ry. For calculating the structural, electronic, and magnetic properties of YN, InN, and YxIn1-xN, a (1×1×2) supercell with 8 atoms was used. Furthermore, Wien2K revealed a tetragonal structure.
3. Results and discussion
3.1 Structural properties of InN, YN, and YxIn1-xN
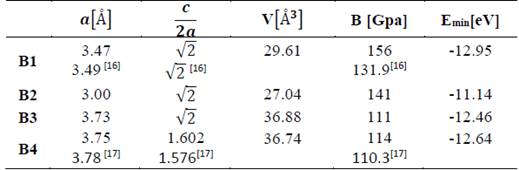
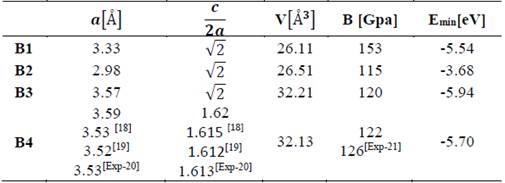
In Tables 1 and 2, the optimized structural parameters are shown, such as: lattice constant (a), ratio (c/a), bulk modulus (B0), and cohesion energy minimum (E0), listed for the YN and InN compounds in the NaCl, CsCl, zincblende, and wurtzite structures, labeled B1, B2, B3, and B4, respectively. Some values of the structural parameters obtained are compared with theoretical and experimental data available in the literature [16-21].
Table 1 The structural parameters of YN in the B1, B2, B3, and B4 structures. Total energies and volumes are given per unit cell (containing 8 atoms). The parameter a0 (Å) in B1, B2, and B3 is taken as where a(Å) is the lattice constant
Source: The Authors
Table 2 Structural parameters of InN in the B1, B2, B3, and B4 structures. Total energies and volumes are given per unit cell (containing 8 atoms). The parameter a0 (Å) in B1, B2, and B3 is taken as a(Å)=a0 (Å)/√2, where a0 (Å) is the lattice constant
Source: The Authors
Table 1 shows the calculations carried out in this investigation and by other authors for YN in the four phases, where the B1 (NaCl) phase and the B4 (WZ) phase are the most energetically favorable structures in the 1×1×2 geometry of YN. They fit well to the theoretical data reported by other authors [16,17]. In Table 2, the data for InN can be seen in the 1×1×2 geometry, calculated in this investigation and by other authors using the 1×1×1 geometry, where it can be seen that the most stable phase is B3 (ZnS phase), with an energy ~0.24 eV higher than the metastable B4 phase (WZ phase) [18-21]. Additionally, for InN in the B4 phase, the lattice parameter, the ratio of c/a, and the modulus of volume are in excellent agreement with the experimental data, ~1.69%, ~0.43 %, and ~3.27 %, respectively.
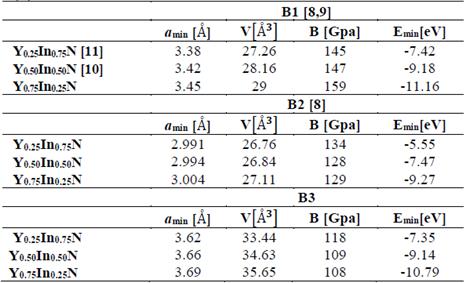
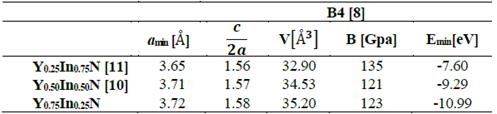
Fig. 1 shows the cohesion energy-volume curves corresponding to the B1, B2, B3, and B4 structures for the different concentrations x of Y atoms considered in this paper. All total energy-volume curves have a parabola-like behavior and were fit to the Murnaghan equation of state [22] in order to determine their ground state properties. In Tables 3 and 4, the structural parameter of YxIn1-xN at the concentrations x = ¼, ½, and – are listed for each of the B1, B2, B3, and B4 structures. It can be seen in Fig. 1 and Table 3 that for the Y0.75In0.25N compound, the B1 structure has a minimum total energy value of ~-11.16 eV/mol, so it can be considered to be the most energetically stable, with differences with respect to the B4, B3, and B2 structures. The least energetically favorable one is the B2 structure, with the highest energy difference with respect to the B1 structure. For Y0.75In0.25N, the B4 structure is the most favorable, with a minimum energy of ~-0.99eV/mol, followed by the B1 structure, with an energy difference of ~0.17eV/mol.
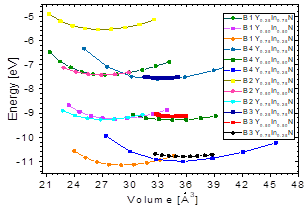
Table 3 The structural parameters of YxIn1-xN in the B1, B2, B3, and B4 structures. Total energies and volumes are given per unit cell (containing 8 atoms). The parameter a0 (Å) in B1, B2, and B3 is taken as a(Å)=a0 (Å)/√2, where a(Å) is the lattice constant, c/2a=√2
Source: The Authors
Finally, for the Y0.75In0.25N compound, the B3 structure is the most favorable one, with a total energy minimum value of ~ -10.79 eV/mol, followed by the B1 structure, with an energy difference of ~0.37eV/mol.(Table 4).
Table 4 Structural parameters of YxIn1-xN in the B4 structure. Total energies and volumes are given per unit cell (containing 8 atoms), where u1(YN) = 0.396 and u2(InN)=0.389
Source: The Authors
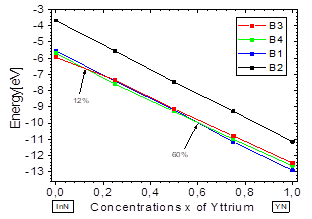
Fig.2 shows the variation in total energy vs. the concentration x of Y, and there it can be seen that for concentrations from x=0 to ~ 0.12, the B3 structure has the lowest energy, and at this point B4 crosses its path, becoming the most stable up to about ~ 0.6, where B1 becomes more stable. These results are in good agreement with previously-reported theoretical and experimental data [2]. They indicate that the YInN compound, as it approaches both ends, adopts the ground structures of the binary compounds B1-B4, belonging to InN and YN.
In Fig. 3, we plot the lattice parameter versus x concentration of Y atoms in the B1, B2, B3, and B4 structures considered in this study. This variation in the four phases is significant and nonlinear, with a downward bowing. The bowing parameters are negative and small 𝑏 𝑎B2 ~-0.01486 Å, 𝑏 𝑎B1 ~-0.08Å, 𝑏 aB3 ~-and b a B4 ~-0.034Å, as can be seen in Table 5. These small values are coherent with the usual assumption that there should be a relative lattice matching in the four phases between the YN and InN binary compounds that causes some strain. This strain may be due to a decrease in the ionic bond length between the cations and the anions as the yttrium replaces indium on the cationic site. Indeed, the cation-anion bond length decrease is usually attributed to the smaller ionic yttrium radius. This suggests that there is a kind of nonlinear charge distribution transferability as the more covalent In-N bond is replaced by a Y-N bond with a more ionic character.
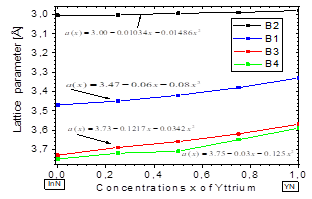
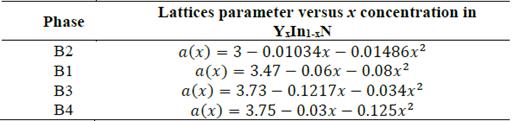
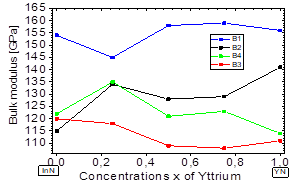
In Fig. 4, it is shown that the bulk modulus decreases in the B1 and B4 phases for values x≥~0.5 of yttrium, and in the B2 and B3 phases it increases for values x≥~0.5 of yttrium. Furthermore, between 0≤x≤~0.25 of yttrium, the bulk modulus decreases in the B1and B4 phases. Also, there is an increase in the B4 and B2 phases, between ~0.5≤x≤1 of yttrium.
3.2 Phase Transition
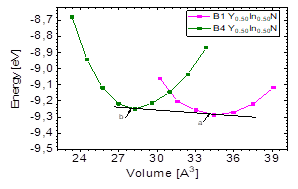
It can be seen in Fig.1 that for the Y 0.50 In 0.50 N compound there is an intersection between the energy-volume curves between the most favorable state, B4, and the B1 state, because there is a phase transition from B4 to B1 caused by pressure [10]. In order to calculate the transition pressure at the cutoff point of Fig. 5, the value of the derivative was graphically calculated at the cutoff point in order to find the value of the transition pressure.
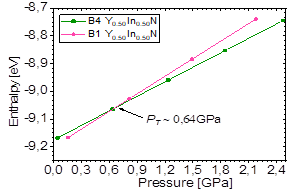
First, a straight line was drawn that passes through the cutoff point, trying to make it pass through some points of the curves, as shown in Fig. 5, where point (a) indicates Y0.50In0.50N in the B1 phase in the conditions near and before the transition from Y0.50In0.50N in the B4 phase, P > P T , and point (b) indicates Y0.50In0.50N in the B4 phase in conditions near and after the phase transition to Y0.50In0.50N in the B1 phase, P < P T . The value of the transition pressure also can be seen more precisely in Fig. 6, where the transition Y0.50In0.50N in the B1 phase to Y0.50In0.50N in the B4 phase is shown. It is predicted that this transition occurs for 𝑃 𝑇 ~0,64𝐺𝑃𝑎; at this pressure, the enthalpies of the two structures are the same.
In the next section, we study the electronic properties of Y0.50In0.50N at the points that are marked in Fig. 5. It is important to determine the electronic properties of this material in its different phases and different volumes in order to study the response of the behavior under pressure. This response is determined by the changes in the distances to first neighbors and changes in symmetry, which are related to the band structure.
3.3 Electronic properties of YInN
In Figs. 7-12, the results are presented for the density of states (DOS) (partial and total) and the band structures for the most stable compound, Y0.75In0.25N in the B1, B4, and B3 phases, respectively. In all the figures, we take the Fermi level as the zero energy.
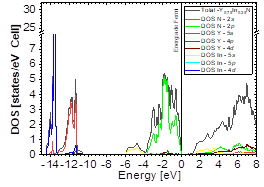
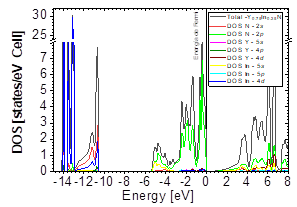
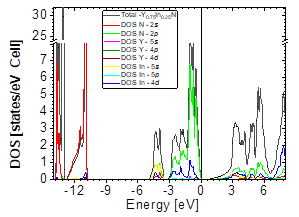
The DOS in Figs. 7-9 shows that the compound exhibits a semiconductor behavior in all three phases. We note that in the valence of the region under ~-10 eV, there are two very high peaks, where their greatest contribution is N-2s and In-4d, and below the Fermi level there exists another peak, where the greatest contribution is from the orbitals N-2p, with small contributions from In-5s. Finally, an energy gap is found at ~0.6eV in the B1 phase and another at ~0.8eV in the B4 phase, while for Y 0.75 In 0.25 N in the B3 phase, it can be seen that in the valence region under ~-10.84eV, there are two very high peaks where the greatest contribution is from N-2s, and below the Fermi level there are three peaks where the greatest contributions are from orbitals N-2p, In-5s, and In-4d, and finally an energy gap of ~0.7eV is noted.
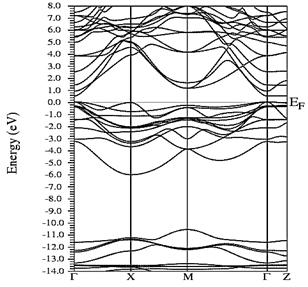
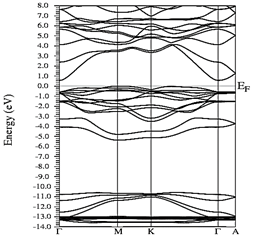
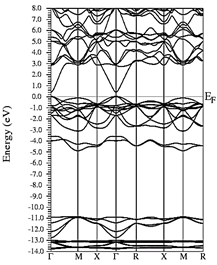
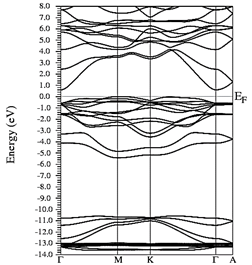
Figs. 10-12 show the band structure of Y0.75In0.25N in the B1, B4, and B3 phases, respectively. From the band structures, Figs. 10-12, we found the same three regions observed in the DOS; in all three, we found a semiconductor band gap. For the B1 phase, there is a direct gap at the Γ point, the B4 has an indirect gap located between the maximum point M in the valence band and the minimum point Γ in the conduction band, and finally the B3 phase has a direct gap located at the Γ point. Finally, Figs. 13-14 and Figs. 15-16 show the DOS and band structure of Y0.50In0.50N in the B1 and B4 phases, respectively. The DOS and the band structure were calculated from points immediately before and after the transition point. In these figures, it can be seen that there are no changes in its structure compared with Y0.75In0.25N in the B1 and B4 phases in Figs. 7-8 and Figs. 10-11. This is because the transition pressure is very small.
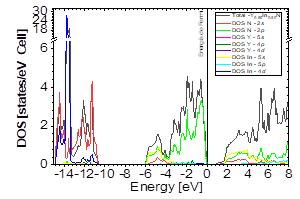
Source: The Authors
Figure 13 DOS partial and total for Y0.50In0.50N in the B1 before of the phase transition to Y0.50In0.50N in the B4.
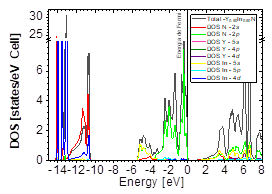
Source: The Authors
Figure 14 DOS partial and total for Y0.50In0.50N in the B4 after of the phase transition Y0.50In0.50N in the B1.
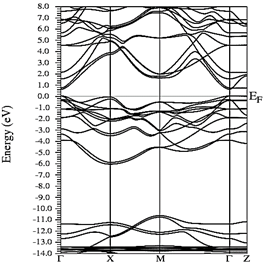
Source: The Authors
Figure 15 Electronic bands for Y0.50In0.50N in the B1 before of the phase transition to Y0.50In0.50N in the B4.
4. Conclusions
We have studied the structural and electronic properties of the YxIn1-xN compound in the B1, B2, B3, and B4 structures for concentrations of x = 0, ¼, ½, –, and 1 with a 1×1×2 geometry, where it was found that these compounds can coexist at a temperature of T=0K. The calculations showed that the Y0.75In0.25N structure in the B1 phase is the most energetically favorable. N. Benyounes et al. suggest that the alloy YInN in the metastable B3 phase allows carrying out the synchronization of the energy gap [5]. In the present investigation, it was found that for the compound in the 1×1×2 geometrical structure between 0<x<(~0.12) x of yttrium, the most stable phase is B3, and a metastable phase in the B4 phase was found with a maximum energy of ~0.24 eV. Furthermore, our calculations show that between ~0.12 <x<~0.6 this compound is more energetically stable in the B4 phase, which is in agreement with that reported by other authors [2] Finally, it was found that between ~0.6 <x<1, the compound is the most stable in the B1 structure, as indicated in previous reports. For the Y0.75In0.25N compound, there is no phase transition between the B2, B3, and B4 structures. We studied the DOS and band structure for the Y0.75In0.25N compound and found that it exhibits a semiconductor behavior, an indirect band gap in the B4 structure, and a direct band gap in the B1 and B3 structures. Finally, for the Y0.50In0.50N compound there is a phase transition from the B4 to the B1 structure, with a volume reduction of ~21% at a transition pressure of ~0.64 GPa. The study of the DOS and band structure for Y0.50In0.50N in the B4 and B1 structures around the points of transition shows no change in the band gap width.

