










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
versión impresa ISSN 0034-7418versión On-line ISSN 1909-6356
Rev. colomb. cienc. quim. farm. v.35 n.1 Bogotá ene./jun. 2006
Artículo de revisión
Integración viral y cáncer de cuello uterino
HPV integration and cervical cancer
Jacqueline López1 y Fabio Ancízar Aristizábal2
1 Grupo de Farmacogenética del Cáncer, Departamento de Farmacia, Universidad Nacional de Colombia, Sede Bogotá, A.A. 14490, Bogotá D.C., Colombia.
2 Instituto de Biotecnología, Universidad Nacional de Colombia, Sede Bogotá,faaristizabalg@unal.edu.co
Recibido para evaluación: marzo 7 de 2006 Aceptado para publicación: junio 15 de 2006
RESUMEN
El cáncer de cuello uterino (CCU) constituye un problema generalizado de salud que ocupa el segundo lugar a nivel mundial entre las neoplasias malignas de la mujer.Las infecciones persistentes con virus del papiloma humano (VPH) de alto riesgo son reconocidas actualmente como un factor necesario en el desarrollo de CCU y sus lesiones precursoras. Uno de los procesos que parece estar más involucrado en el origen de las células malignas, es el evento de integración del virus al genoma del huésped, relacionado también con la progresión de lesiones preinvasivas y el mantenimiento del fenotipo transformado. Estos eventos de integración, especialmente de VPH tipo 16 se dan en secuencias específicas del genoma viral, principalmente en su región E1/E2, interrumpiendo la secuencia y permitiendo que exista desregulación de las actividades de transcripción, conduciendo a la sobreexpresión de las oncoproteínas virales E6 y E7 que normalmente inactivan genes supresores de tumores, como p53 y pRb, responsables del control en importantes puntos de chequeo del ciclo celular. Actualmente el proceso de integración viral, es considerado como una alteración genética importante que caracteriza las displasias malignas, con potenciales aplicaciones como marcador de progresión de lesiones precursoras y herramienta de diagnóstico.
Palabras clave: virus del papiloma humano, integración viral, cáncer de cuello uterino, lesiones intraepiteliales de alto grado.
SUMMARY
Cervical cancer is the second most prevalent cancer worldwide and is the second leading cause of cancer deaths in women. Persistent infection with high-risk human papillomaviruses (HPVs) types are the causative agents of cervical cancer, since 99% of tumors are positive for HPV DNA, HPV is a necessary cause of invasive cervical cancer worldwide. One of the key events of HPV-induced carcinogenesis is the integration of the HPV genome into a host chromosome, related with precancerous lesions progression. Integration follows a more specific pattern with respect to the HPV genome, expression of the viral E6 and E7 genes is consistently maintained, whereas other positions of the viral DNA are deleted or their expression is disturbed, like E2 gene. Loss of expression of the HPV E2 transcriptional repressor is significant and may be critical for malignant progression, as it may result in increased HPV E6 and E7 expression. The HPV E6 and E7 oncoproteins inactivate the p53 and pRB tumor suppressors; expression of high-risk HPV E6/E7 oncogenes provides a subset of the minimally required carcinogenic hits for full transformation of human epithelial cells. There is also evidence for deregulated HPV-16 E6-E7 mRNA stability after integration and specific alterations of host cellular gene expression have been detected upon HPV genome integration. Currently, integration of HPV is considered to be an important genetic alteration for the progression of intraepithelial lesions to invasive disease with potential clinical applications like mark tumour progression and diagnostic tool.
Key words: human papillomavirus, viral integration, cervical cancer, high grade squamous intraepithelial lesions.
INTRODUCCIÓN
El cáncer de cuello uterino (CCU) es el segundo cáncer más frecuente a nivel mundial después del cáncer de seno, constituyéndose en uno de los principales problemas de salud pública en la población femenina (1). En muchos países en desarrollo, especialmente en África y América Latina, ocupa el primer lugar en incidencia; sin embargo, es una enfermedad que a pesar de sus altas tasas de incidencia y mortalidad, puede ser prevenida cuando es detectada en sus etapas iniciales (2).En Colombia, el CCU es la principal causa de mortalidad por cáncer en edad reproductiva debido a que la enfermedad es detectada en estadios avanzados en un número importante de mujeres y a la resistencia a la radioterapia (3). Según datos de la Agencia Internacional de Investigación en Cáncer (IARC), para el año 2002 la patología presentó una incidencia anual de 36,4 nuevos casos por cada 100.000 mujeres y una tasa demortalidad de 18,2/100.000, valores superiores al promedio de los países latinoamericanos.
El CCU, al igual que muchos otros cánceres, se desarrolla como resultado de múltiples alteraciones genéticas, epigenéticas e inmunológicas; adicionalmente otros factores como la interacción entre medio ambiente y célula pueden facilitar la promoción de los procesos de transformación neoplásica. Una de dichas interacciones la constituye la infección con el virus del papiloma humano (VPH) considerado, según la historia natural de la ontogénesis de cuello uterino, como el principal factor etiológico de este tipo de cáncer. Numerosos estudios epidemiológicos han establecido la relación causal entre la infección viral y el desarrollo de neoplasias de cuello uterino (2,4,5). El patógeno que es detectado en el 90-100% de los casos, se cree es una causa necesaria pero insuficiente para el desarrollo de carcinomas invasivos de cuello uterino (CIC) (6).
Una gran variedad de VPH han sido identificados, pero solo los tipos de alto riesgo (principalmente 16 y 18) son asociados con cáncer de cuello uterino, debido a su gran capacidad transformante y a su frecuente presencia en lesiones neoplásicas avanzadas.Los virus de bajo riesgo se encuentran principalmente en lesiones intraepiteliales de bajo grado (LIEBG), atipias que en la mayoría de los casos regresan espontáneamente sin ocasionar problemas en el tejido al que infectan.
Uno de los procesos que parece estar más involucrado en el origen de las células malignas es el evento de integración del virus al genoma del huésped, proceso actualmente considerado como una alteración genética importante que caracteriza las lesiones malignas y que se relaciona con la pérdida o inactivación de genes.Estos eventos de integración, mejor caracterizados en el tipo viral 16, se dan en sitios específicos de la región E1-E2 interrumpiendo la secuencia y permitiendo la desregulación de las actividades de transcripción, resultando finalmente en la sobreexpresión de las oncoproteínas virales. Las proteínas E6 y E7 normalmente inactivan genes supresores de tumores, como p53 y pRb, conduciendo al descontrol en importantes puntos de chequeo del ciclo celular y de esta manera, favorecen la progresión de lesiones preinvasivas y el mantenimiento del fenotipo maligno (7).Por tanto, el evento de integración del virus en el genoma del huésped ha sido propuesto como potencial marcador de la progresión de neoplasias cervicales.
Teniendo en cuenta que el pronóstico de pacientes con CCU es generalmente pobre mientras que las lesiones precursoras son frecuentemente curables, el conocimiento de los eventos moleculares que hacen que una lesión progrese es de considerable interés debido a la necesidad de identificar marcadores moleculares que permitan definir si la lesión va a progresar y en esta medida, darle un tratamiento más adecuado al paciente. La detección de las tasas de integración contribuirá de manera directa y efectiva en la toma de decisiones para el manejo y seguimiento de pacientes con lesiones intraepiteliales, disminuyendo de esta manera el riesgo de CCU.
VIRUS DEL PAPILOMA HUMANO
La infección por VPH es considerada como la enfermedad de transmisión sexual más frecuente a nivel mundial, existen estimaciones que la ubican hasta en el 80% de las mujeres asintomáticas con vida sexual activa. Dentro de la etiología multifactorial que caracteriza el desarrollo de CCU se destaca la participación primordial que tiene la infección por VPH. Actualmente, la infección persistente con genotipos de alto riesgo, principalmente VPH 16 y 18, ha sido reconocida como la causa central y virtualmente necesaria en el desarrollo, mantenimiento y progresión de lesiones premalignas a CCU (6). En efecto, la asociación etiológica entre la infección viral y el CCU es ampliamente aceptada y justificada por numerosos estudios epidemiológicos multicéntricos (2, 4, 5, 8).
Aunque la presencia de VPH es considerada como el principal factor de riesgo, no todas las infecciones con tipos de alto riesgo conducen al desarrollo del fenotipo maligno. La infección por VPH es muy común principalmente en las mujeres jóvenes, que en la mayoría de los casos se resuelve espontáneamente; otras infecciones conducen al establecimiento de lesiones precursoras, de las cuales solamente una pequeña fracción progresa a carcinomas de cuello uterino. El hecho de que sólo una minoría de infecciones persistentes por VPH progrese a cáncer, señala la coexistencia de otros cofactores que deben estar presentes para que se dé la transformación.En otras palabras, la infección es causa central pero insuficiente en la carcinogénesis cervical.
El VPH es un virus relativamente pequeño, sin envoltura lipídica, constituido por ADN circular de doble cadena con una longitud aproximada de 8.000pb.El genoma está dividido en región codificante que comprende ocho genes transcritos en un mARN policistrónico y una región no codificante. La región no codificante o reguladora Long Control Region, (LCR) contiene el origen de replicación, una región promotora (P97 en VPH16) y una secuencia intensificadora que controlan la trascripción de los genes virales. La región codificante está dividida a su vez en dos regiones de expresión, temprana y tardía. Los primeros genes (E1, E2, E4, E5, E6 y E7) están relacionados con la modulación de los procesos de transformación celular, replicación y transcripción viral. La región de expresión tardía contiene los genes L1 y L2 que codifican para las dos proteínas estructurales de la cápside (Figura 1) (1).
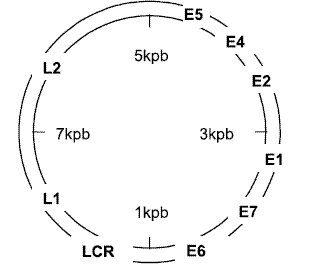
Los productos de expresión de los genes E6 y E7 son considerados elementos claves en la ontogénesis de cuello uterino. Ampliamente se ha documentado la participación de E6 y E7 en la inmortalización de queratinocitos primarios in vitro y su actividad tumorogénica in vivo en micos transgénicos. Adicionalmente, numerosos estudios bioquímicos han clarificado algunos mecanismos de acción de las proteínas multifuncionales E6 y E7, dada su capacidad de formar complejos estables con diferentes proteínas celulares y alterar total o parcialmente sus funciones normales.Las oncoproteínas se unen a proteínas reguladoras del ciclo celular, p53 y pRB, e interfieren en los puntos de chequeo G1/S y G2/M, induciendo diferentes cambios como alteraciones cromosómicas numéricas (1,5).
Taxonómicamente el virus del papiloma humano pertenece a la familia Papillomaviridae, compuesta por 16 géneros. Los tipos virales del género alpha papillomavirus se caracterizan por ser causantes de lesiones mucosas y cutáneas en humanos y primates. Los alpha papillomavirus se han clasificado en dos categorías, virus de alto riesgo (tipos 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 73 y 82) y virus de bajo riesgo (tipos 6, 11, 40, 42, 43, 44, 54, 61, 70, 72 y 81). Los virus de bajo riesgo se han asociado a enfermedades de cuello uterino benignas como condilomas acuminados (verrugas genitales) mientras que los virus de alto riesgo o tipos virales oncogénicos son causantes de displasias severas y CCU (8).
Actualmente, la clasificación de los diferentes tipos virales se realiza por comparación de la secuencia nucleotídica del gen L1, una de las regiones más conservadas dentro del genoma viral. Un nuevo tipo viral se define cuando se identifica un nivel de variación mayor al 10% en la secuencia de L1 respecto a la secuencia de los tipos previamente clasificados, un subtipo entre el 10 y 2% y una variante menos del 2%. De la misma manera, miembros de diferentes géneros comparten menos del 60% de homología en el marco abierto de lectura L1; especies de un mismo género, entre el 60-70%, y tipos de una misma especie, entre el 71-89%. Igualmente, a partir de las sustituciones nucleotídicas presentes en diferentes regiones del genoma viral, como L1, L2, E6 y LCR, se han definido cinco grupos filogenéticos o variantes: Europeo (E), Asiático (A), Asiático-Americano (AA), Africano 1 (Af1) y Africano 2 (Af2) (9).
INTEGRACIÓN VIRAL
El ADN viral puede encontrarse en la célula infectada de tres maneras, integrado al genoma celular (forma integrada), libre en el núcleo celular (forma episomal) y compartiendo las formas integrada y episomal (forma mixta).
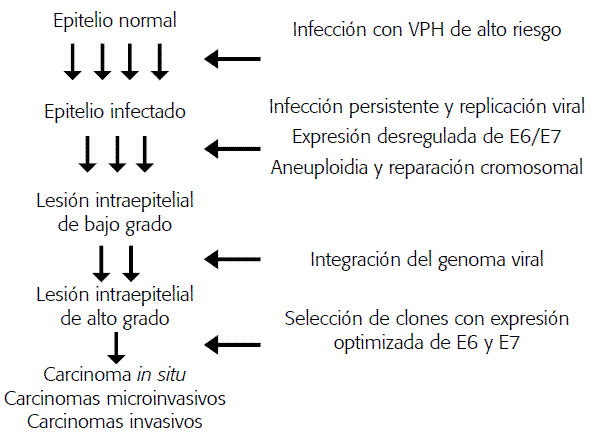
Diferentes trabajos han demostrado que la integración viral es un evento importante en la progresión de lesiones intraepiteliales a carcinomas invasivos de cuello uterino, sin embargo, después de más de dos décadas de investigación en VPH no son claros los mecanismos moleculares involucrados en el proceso de integración. Recientemente, Wentzensen (10) propuso un modelo que intenta explicar el papel de la integración en el desarrollo de carcinomas invasivos (Figura 2). En etapas tempranas, la expresión de oncoproteínas virales en células epiteliales generalmente conduce al desarrollo de diferentes tipos de anormalidades cromosomales (11), no obstante, la posterior activación de los mecanismos de reparación celular facilita la inserción de secuencias foráneas como las de VPH (12).Así, se ha observado que la homología entre secuencias del virus y del genoma celular que se recombinan es prácticamente nula, lo que permite considerar el proceso de inserción viral como un mecanismo que se facilita por eventos de recombinación no homóloga, proceso empleado frecuentemente por las células en la reparación de daños genéticos (13,14). La progresión de lesiones asociadas a VPH refleja el clásico escenario de selección en el cual ciertos eventos conducen a la expansión clonal de células alteradas biológicamente. Pese a que la integración de fragmentos virales ocurre paralelamente en múltiples clones celulares, el proceso de selección en etapas avanzadas promueve el crecimiento de clones celulares que por diferentes mecanismos, como el evento de integración, presentan aumento en la expresión de las oncoproteínas virales (15).
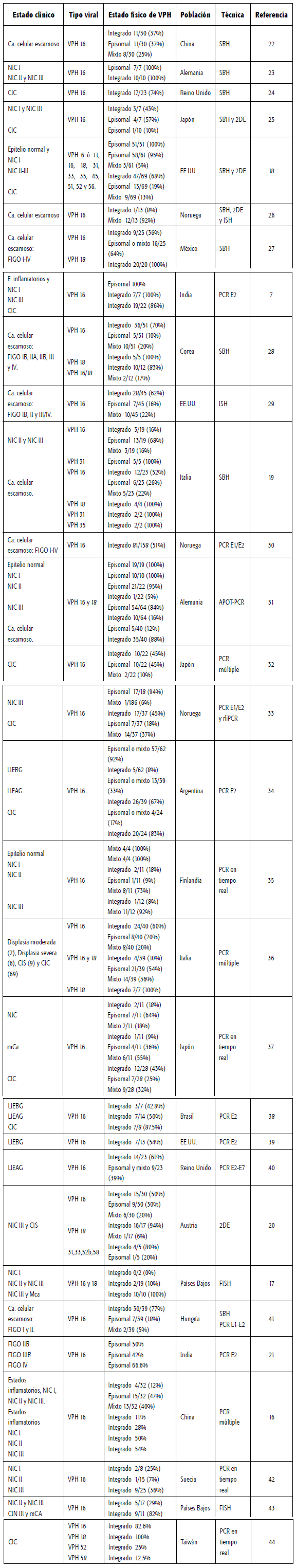
De acuerdo con los resultados resumidos en la tabla 1, la frecuencia de formas integradas o mixtas aumenta con la progresión de la enfermedad de cuello uterino (16); de esta manera, la transición de lesiones intraepiteliales de alto grado (LIEAG) a carcinomas microinvasivos (mCA) se caracteriza por la integración de secuencias VPH de alto riesgo (17). Los resultados experimentales demuestran que la integración es común en CIC y poco frecuente en epitelios normales y neoplasias intraepiteliales de cuello uterino grado I (NIC I), mientras que en NIC II y NIC III la integración viral abarca un amplio rango de valores. Sin embargo, la ausencia de secuencias integradas VPH 16 en algunos estados clínicos avanzados como NIC III o carcinoma in situ (CIS) sugiere que la integración no es siempre requerida para la progresión maligna de lesiones de cuello uterino (15, 18, 19, 20, 21).
SITIOS DE INTEGRACIÓN VIRAL Y SU EFECTO
En el proceso de integración, la linearización del genoma circular es una etapa necesaria que involucra el rompimiento de la doble cadena viral principalmente en la región E1/E2 (24, 29, 30, 33, 45-47). Adicional al rompimiento, en la mayoría de casos se presentan deleciones de fragmentos virales (22, 46).En VPH 16 se han identificados diferentes patrones de ruptura de acuerdo a la secuencia viral afectada, actualmente se considera que el fragmento viral correspondiente a los genes E1 y E2 es la región con mayor tasa de rompimiento. Sin embargo, dentro de esta región a su vez existen varias secuencias específicas que han sido reconocidas como blancos de rompimiento viral tanto en E1 (15, 46, 48) como en E2 (29, 33). Es posible que así como la dinámica de integración varíe con la población de estudio, ocurra lo mismo con los sitios específicos de corte viral.
Con relación a las deleciones, en VPH 16 se han registrado pérdidas en la región E2 de 251 pb (49) y 813 pb (50), deleciones totales en E4 y E5 y parciales en L1 y L2 de 376 pb (51) y en E1 de 1464 pb (52). En VPH 18 aproximadamente la mitad del genoma viral se pierde en la integración comprometiendo total o parcialmente los genes E2, E4, E5, L1 y L2 (47).De otro lado, se han identificado regiones virales que no son blancos de rompimientos en el proceso de inserción como las correspondientes a los genes E6, E7 y LCR (24, 45, 51, 52).
Teniendo en cuenta que frecuentemente ocurren rupturas y/o deleciones a nivel de la región E1 y E2, parte de los efectos de la integración viral van a depender de la alteración de las funciones biológicas de E1 y E2.La proteína E2 de VPH 16 controla negativamente la transcripción de los genes virales E6 y E7 al unirse al promotor viral P97. En presencia de la proteína E2 hay represión continua de la transcripción de las oncogenes E6 y E7, contrariamente, la ausencia del represor E2 permite el libre acceso de factores de trascripción celulares que se agrupan en el promotor viral y regulan positivamente la transcripción de E6 y E7 (53). De acuerdo con lo anterior, recientemente se evidenció que la integración de VPH 16 asociada con deleciones en el extremo 3 de E2 conduce a la desregulación del promotor P97 y al aumento en los niveles de E7 (54).
La pérdida de la función represora de E2 es un factor crítico en la progresión maligna, in vitro se ha demostrado que la reintroducción de E2 en líneas celulares de carcinoma de cuello uterino causa supresión del crecimiento debido a la represión de la transcripción de E6 y E7 (55-58). Igualmente es posible revertir el efecto causado por E2 a través de la reintroducción de las oncoproteínas E6 y E7.Estos experimentos demuestran claramente que la continua expresión de E6 y E7 es necesaria para el mantenimiento del fenotipo maligno y que la expresión de los oncogenes es afectada por la actividad de E2 (57).
La proteína E2 también ha sido relacionada con procesos de muerte celular por apoptosis en ausencia de factores de crecimiento (59).Es interesante anotar que las proteínas E2 de tipos virales de alto riesgo localizadas en núcleo y citoplasma pueden inducir apoptosis, mientras que las derivadas de genotipos de bajo riesgo ubicadas exclusivamente en el núcleo no causan muerte celular. Recientemente se demostró que la acumulación de E2 VPH 16 y 18 en el citoplasma induce apoptosis por activación de la ruta caspasa 8 (60).
Otras consecuencias de la pérdida de la actividad represora de E2 son el aumento en la capacidad de inmortalización (61), el incremento en la estabilidad de los mensajeros E6 y E7 VPH 16 (62) y la alteración del perfil de expresión de varios genes celulares (63). Igualmente, la expresión de E6 y E7 a partir de secuencias integradas confieren a las células infectadas ventajas selectivas de crecimiento (64).
El rompimiento del gen viral E2 ha sido asociada con la progresión de lesiones de cuello uterino, particularmente de LIEBG a LIEAG (34), con bajas tasas de sobrevida y con pronósticos desfavorables en pacientes con CCU asociado a VPH (29, 30, 65); no obstante la presencia de regiones E2 inalteradas junto con la expresión concomitante de oncoproteínas virales en algunos casos de lesiones avanzadas plantea que la ruptura de la secuencia viral en E2 no es siempre un paso indispensable en el desarrollo de CCU, ya que la alteración de la función biológica de E2 puede darse por otros mecanismos alternos tales como cambios en la secuencia nucleotídica (19, 21).
INTEGRACIÓN Y ONCOPROTEÍNAS VIRALES
En carcinomas de cuello uterino la mayoría de genomas VPH integrados son transcripcionalmente activos y expresan oncoproteínas virales, debido a la pérdida de la actividad represora de E2 (15, 21). Sin embargo, pueden existir muchas copias VPH integradas y solamente un bajo porcentaje, menor al 10% en VPH 16, pueden ser transcripcionalmente activas (66, 67).
Efectivamente, diferentes trabajos han demostrado que la inserción de genotipos de alto riesgo en el genoma de la célula huésped está asociada con el aumento en los niveles de transcripción de los genes E6 y E7 (20, 28, 62, 64). Esta observación también ha sido corroborada in vitro, empleando una línea celular derivada de una lesión intraepitelial de bajo grado, en donde las secuencias VPH integradas sobreexpresaron E7 y no generaron transcritos para E2 (54).
Si se considera que en lesiones avanzadas prevalecen las formas integradas y que E2 es un represor transcripcional de los oncogenes virales, es posible predecir que en displasias severas y en CIC se presente mayor expresión de E6 y E7. Recientemente se demostró la ausencia de mensajeros para las oncoproteínas virales en todos los casos de NIC I y NIC II y la presencia de mARN en la totalidad de los casos de NIC III y CIC, estos resultados sugieren la posibilidad de emplear la valoración de los niveles de mARN de E6 y E7 como un indicador de pronósticos en pacientes con CCU asociado a VPH (21).
Teniendo en cuenta que tanto el evento de integración como la presencia o ausencia de mensajeros E6/E7 han sido relacionados con la progresión de la enfermedad y que el número de copias virales aumenta en LIEAG, Rajeevan (67) evaluó la relación entre los niveles de transcritos y la carga viral en NIC II y NIC III. El trabajo concluye que aunque en todos los casos analizados se observó una correlación positiva entre los niveles de mensajeros y la carga viral, ésta fue independiente del estado clínico, es decir, no se presentaron diferencias en los niveles de mensajeros entre NIC II y III.Aunque la inalteración de esta relación en diferentes estados clínicos no excluye que la desregulación de las oncoproteínas virales sea un mecanismo biológicamente significativo en la progresión maligna, son necesarios más adelantos en este sentido antes de considerar la evaluación de los niveles de mensajeros E6/E7 como posibles marcadores de pronóstico.
SITIOS DE INTEGRACIÓN CELULAR Y SU EFECTO
El proceso de inserción viral no solo afecta la estructura del genoma viral sino también los genes celulares del hospedador interrumpidos durante la integración. Actualmente se han identificado más de 230 sitios de integración celular, la mayoría correspondientes al tipo viral 16 descritos en tumores primarios de cuello uterino y líneas celulares (13-15, 33, 35, 48, 52, 68-86).
La integración de secuencias VPH ocurre dentro de regiones del genoma de baja condensación, transcripcionalmente activas, que permite mayor accesibilidad para la integración de ADN foráneo (87). Aunque actualmente es aceptada la hipótesis en donde se plantea que el evento de integración viral ocurre aleatoriamente a lo largo del genoma, un alto porcentaje de los sitios celulares de integración corresponden a CFSs (Common Fragile Sites) o regiones genómicas altamente susceptibles a la ruptura que facilitan la inserción de ADN exógeno. La integración en CFSs es comúnmente asociada con grandes deleciones cromosomales y rearreglos, por tanto cualquier gen o grupo de genes presentes en estas regiones podrían sufrir este tipo de alteraciones después de la integración de secuencias exógenas (83).
Se ha observado integración de fragmentos virales en genes involucrados con el desarrollo de tumores como MYC, TP63, NR4A2, APM-1, FANCC, TNFAIP2 y hTERT.A pesar de ser interrumpidas frecuentemente las regiones intrónicas en el proceso de integración, la estructura de los mensajeros, y por ende la expresión de genes, puede alterarse significativamente. En el caso de MYC, dos trabajos evidenciaron que la integración de VPH conduce al aumento de los niveles de mensajero del oncogen celular, sugiriendo fenómenos de activación debidos al proceso de integración (68,72). De otro lado, uno de los cuatro casos de integración estudiados en la región promotora de hTERT presentó aumento en el nivel de transcripción e incremento marcado de la actividad telomerasa (84,88), sin embargo, la activación de la enzima telomerasa es un evento frecuente en cáncer de cuello uterino asociado a VPH ya que la proteína E6 produce transactivación de la transcripción de hTERT (89). En general, existe poca evidencia experimental que relacione la integración viral con la alteración de la actividad biológica de los genes celulares afectados y por tanto no es clara la contribución de estas modificaciones celulares en el desarrollo del fenotipo maligno.
A partir del análisis de tumores asociados a VPH y del desarrollo de un modelo de integración viral in vitro, se ha planteado una nueva hipótesis que establece la continua inserción de secuencias virales en células epiteliales infectadas, sin embargo, solamente algunas integraciones específicas podrían alterar los genes celulares localizados en los sitios de integración o sus alrededores, este evento junto con el aumento del nivel de transcritos de oncoproteínas virales mediado por E2 contribuiría conjuntamente en el desarrollo de tumores de cuello uterino (86).
INTEGRACIÓN VIRAL Y ESTABILIDAD CROMOSOMAL
La sobreexpresión de oncogenes E6 y E7 es el principal factor viral en la patogénesis de displasias de cuello uterino. Actualmente se reconoce que la expresión de los genes E6 y E7 en células epiteliales conduce al desarrollo de inestabilidades cromosomales e induce aneuploidía, así mismo la pérdida de la estabilidad genética del huésped favorece el evento de integración lo que a su vez resulta en la sobreexpresión de E6 y E7. Se ha demostrado in vivo que la aneuploidización precede el evento de integración viral en la progresión de displasias de cuello uterino (12).Las regiones cromosomales donde se presenta mayor incidencia de imbalances así como un alto número de amplificaciones, coinciden con CFSs y otros locis celulares descritos como sitios de integración celular (90, 91).
Recientemente se observó una correlación significativa entre el número de copias cromosomales y el estado físico viral, las formas episomales inducen tetrasomias y trisomias mientras que las formas integradas se relacionan con el desarrollo de aneusomias y polisomias, detectadas predominantemente en NIC III y carcinomas microinvasivos (17). De la misma manera se observó mayor frecuencia de imbalances numéricos en muestras que presentaron pérdida de la región E2 en el genotipo 16 (40).La sobreexpresión de E7, por disrupción de E2, es asociada con diferentes formas de inestabilidad genómica, en estados donde coexisten las formas integradas y episomales los niveles intermedios de E7 son relacionados con anormalidades numéricas mientras que en formas integradas, los altos niveles de E7 correlacionan con anormalidades numéricas y estructurales (54).
INTEGRACIÓN Y OTROS FACTORES VIRALES
Dentro de los factores de riesgo virales asociados con CCU, el genotipo, las variantes intratípicas y la carga viral han sido relacionados con la integración de secuencias VPH.
La mayoría de estudios de integración viral se han realizado en los dos principales genotipos de alto riesgo, VPH 16 y VPH 18. Comparando las tasas de integración entre estos genotipos es posible concluir que el comportamiento de los tipos 16 y 18 difiere ampliamente (tabla 2), la inserción de secuencias VPH 18 parece ser un evento crucial y más eficaz en la transformación de displasias a carcinomas y se asocia con un mayor grado de agresividad en términos de rápida progresión maligna. No obstante, tanto la tipificación viral como la evaluación del estado de integración son parámetros que permitirían un diagnóstico más acertado y un mejor pronóstico de displasias de cuello uterino (20, 36). En infecciones múltiples VPH 18 y 16, el genotipo 16 presenta alto porcentaje de formas episomales respecto a la infección exclusiva por VPH 16, planteándose la posibilidad de competencia entre los dos tipos virales por los sitios de integración celular (36).

En cuanto a las variantes intratípicas, es interesante anotar que dos trabajos desarrollados en la población mexicana identificaron varias sustituciones nucleotídicas en E2 concomitante con el mantenimiento de la integridad física del gen en la mayoría de carcinomas evaluados, los resultados sugieren que la variación genética de la región E2 puede ser un mecanismo alterno de desregulación en la expresión de oncoproteínas virales. Efectivamente, las diferencias en la regulación de la transcripción de E6 y E7 por la proteína E2 pueden ser asociadas con una mayor oncogenicidad de las variantes asiático-americanas (AA), presentes mayoritariamente en la población latinoamericana (92, 93). Contrario a lo observado con las variantes AA, las variantes europeas (E) se asociaron con la alteración física del gen E2 sugiriendo que la sobreexpresión de E6 y E7 en variantes E inicia en etapas tardías de la infección viral, ya que es indispensable la ruptura de la secuencia viral y su posterior inserción en el genoma del huésped (94). Es importante resaltar que la modificación del estado de metilación del genoma de VPH, al igual que las sustituciones nucleotídicas, puede conducir a la alteración de la funcionalidad de genes virales; así, el incremento en el nivel de metilación del gen L1 y la disminución en E6 y LCR, de VPH 16 y 18, han sido asociados con etapas avanzadas del CCU. A la fecha, aunque no existe suficiente evidencia experimental relacionada con la dinámica de metilación de otros genes virales, como E2 y E1, es posible que la variación en estos niveles conduzca a la alteración de la actividad biológica (95-98).
Teniendo en cuenta que la carga viral es un valor predictivo de persistencia y progresión (99-101) y que la rápida progresión de neoplasias intraepiteliales ha sido asociada con una alta carga de secuencias integradas (35), es posible considerar la identificación del estado de integración junto con la determinación de la carga viral como parámetros de pronóstico.Sin embargo, son pocos y no concluyentes los trabajos que correlacionan integración y carga viral. Berumen (27) y Jeon (64) asociaron la integración de secuencias VPH 16 con un menor número de copias virales, mientras que Anderson (42) demostró que el promedio de número de copias de VPHs de alto riesgo en muestras NIC I a III no presentó diferencias entre los tres estados clínicos evaluados, en donde en la mayoría de casos se observó integración. Es posible que la dificultad en esta clase de trabajos radique en la amplia variedad de técnicas desarrolladas para la valoración de la carga viral e identificación del estado físico viral, cada procedimiento con limitantes técnicas y conceptuales específicas. En este sentido es necesaria la ejecución de trabajos orientados a verificar la sensibilidad y especificidad de las técnicas así como el grado de concordancia entre ellas.
TÉCNICAS DE DETECCIÓN
Aunque han sido desarrolladas diferentes técnicas cualitativas y cuantitativas (tabla 3), la PCR es la más empleada en la identificación del estado físico de VPH. Inicialmente la categorización por PCR se basaba en la amplificación del gen E2, la ausencia o presencia de producto amplificado representaba las formas integradas y episomales respectivamente; sin embargo, esta técnica no permitía discriminar las formas episomales de las mixtas ya que en ambos casos se obtenía producto amplificado. Actualmente, la discriminación de los tres estados físicos por PCR múltiple y en tiempo real se basa en la cuantificación de la relación E2/E6; en formas integradas puras no se observan amplímeros correspondientes a E2, en formas episomales puras la relación de E2 y E6 es equivalente, mientras que en formas mixtas se genera menor señal para E2 respecto a E6.
Una de las razones que explica la alta variabilidad de los resultados en NIC II y NIC III es el empleo de diferentes técnicas y procedimientos en la detección del estado físico viral y más aún, la ausencia de trabajos que reporten el grado de concordancia entre éstas. Recientemente se evaluaron estados de integración en muestras de NIC y CIC empleando PCR en tiempo real y FISH, los datos demostraron concordancia de formas integradas en 37 de 43 casos evaluados (86%), concluyendo que las dos técnicas no solo fueron convenientes sino complementarias en la evaluación de tasas de integración (105).
A pesar de los constantes adelantos en el mejoramiento de los procedimientos por ISH y PCR, deben tenerse en cuenta varias limitantes técnicas en la interpretación de resultados. Al emplear PCR, debería analizarse la totalidad de la región E1/E2, debido a los múltiples patrones de ruptura reportados en esta región, sin embargo, en muchos casos se han seleccionado secuencias cortas de corte que como se mencionó anteriormente, posiblemente pueden variar de acuerdo a la población de estudio. En el caso de FISH, un bajo número de copias episomales y la presencia de formas concaterizadas pueden generar señales puntuales, es decir, falsos positivos de integración (106).
Aunque actualmente existen plataformas tecnológicas que permiten el análisis masivo de muestras tanto por PCR como por FISH, es evidente que hay una tendencia hacia el empleo de técnicas basadas en PCR, no sólo por costos y tiempo de análisis sino porque a diferencia de FISH, su uso en numerosos estudios ha permitido corroborar la validez de los resultados.
PERSPECTIVAS
La determinación de las tasas de integración, como potencial marcador molecular de progresión, es un tema de alta importancia médica en el orbe y particularmente en Colombia si se tiene en cuenta que el cáncer de cérvix es la principal causa de mortalidad por cáncer. Aunque han sido implementadas diferentes técnicas para la detección de las formas físicas de VPH, son evidentes las múltiples limitaciones metodológicas que en muchos casos ponen en duda la veracidad de varios estudios; a la fecha, las investigaciones asumen que la ruptura viral ocurre con frecuencia en E2 y aún más, limitan el análisis a un pequeño segmento del gen. Es claro que aún falta investigación encaminada a validar las metodologías empleadas en la detección de las formas físicas de VPH, así como estudios que evalúen el grado de concordancia entre éstas. No obstante a futuro, la validación de las tasas de integración como marcador molecular de progresión contribuirá de manera directa y efectiva en la toma de decisiones para el manejo y seguimiento de pacientes con lesiones intraepiteliales, disminuyendo de esta manera el riesgo de CCU.
AGRADECIMIENTOS
Al Posgrado Interfacultades en Microbiología y a la Facultad de Medicina de la Universidad Nacional de Colombia por su colaboración durante la consulta bibliográfica.
BIBLIOGRAFÍA
1. M.S. Longworth y L.A. Laimins,Pathogenesis of human papillomaviruses in differentiating epithelia, Microbiol.Mol. Biol. Rev., 68, 362 (2004). [ Links ]
2. F.X. Bosch, A. Lorincz y N. Muñoz, Thecausal relation between human papillomavirus and cervical cancer, J. Clin. Pathol.,55, 244 (2002). [ Links ]
3. InstitutoNacional de Cancerología. "Anuario Estadístico 2004", Bogotá, 2005. [ Links ]
4. N. Muñoz,Human papillomavirus and cancer: the epidemiological evidence, J. Clin. Virol., 19, 1 (2000). [ Links ]
5. F.X. Bosch y S. Sanjosé, Humanpapillomavirus and cervical cancer-burden and assessment of causality, J. Nat. Cancer. Inst. Monogr., 31, 3 (2003). [ Links ]
6. J.M. Walboomers, M.V. Jacobs y M.M.Manos, Human papillomavirus is a necessary cause of invasive cervical cancerworldwide, J. Pathol., 189, 12 (1999). [ Links ]
7. B. Daniel, G. Mukherjee y L. Seshadri,Changes in the physical state and expression of human papillomavirus type 16 in the progression ofcervical intraepithelial neoplasia lesions analysed by PCR, J. Gen. Virol., 10, 2589 (1995). [ Links ]
8. N. Muñoz, F. Bosch y S. Sanjosé,Epidemiologic classification of human papillomavirus types associated withcervical cancer, N. Engl. J. Med., 348, 518 (2003). [ Links ]
9. E. de Villiers, C. Fauquet y T.R.Broker, Classification of papillomaviruses, Virol., 324, 17(2004). [ Links ]
10. N. Wentzensen, S. Vinokurova, M. vonKnebel-Doeberitz, Systematic review of genomic integration sites of humanpapillomavirus genomes in epithelial dysplasia and invasive cancer of thefemale lower genital tract, Cancer. Res., 64, 3878 (2004). [ Links ]
11. S.Duensing y K. Munger, The human papillomavirus type 16 E6 and E7 oncoproteinsindependently induce numerical and structural chromosome instability, Cancer.Res., 62, 7075 (2002). [ Links ]
12. P.Melsheimer, S. Vinokurova y S. Wentzensen, DNA aneuploidy and integration ofhuman papillomavirus type 16 E6/E7 oncogenes in intraepithelial neoplasia andinvasive squamous cell carcinoma of the cervix uterine, Clin. Cancer. Res., 10, 3059 (2004). [ Links ]
13. M.H.Einstein, Y. Cruz y M.K. el-Awady, Utilization of the human genome sequencelocalizes human papillomavirus type 16 ADN integrated into the TNFAIP2 gene ina fatal cervical cancer from a 39-year-old woman, Clin. Cancer. Res., 8,549 (2002). [ Links ]
14. E.C.Thorland, S.L. Myers y D.H. Persing, Human papillomavirus type 16 integrationsin cervical tumors frequently occur in common fragile sites, Cancer. Res., 60, 5916 (2000). [ Links ]
15. C.Ziegert, N. Wentzensen y S. Vinokurova, Comprehensive analysis of HPVintegration loci in anogenital lesions combining transcript and genome-basedamplification techniques, Oncogene.,22, 3977 (2003). [ Links ]
16. Q.Y.Chen, M.L. Bian y Z.H. Chen, [Detection of integration status of humanpapillomavirus 16 incervical precancerous lesions], Zhonghua Yi Xue Za Zhi, 85, 400 (2005).Resumen consultado en inglés. [ Links ]
17. A.H. Hopman, F. Smedts y W.Dignef, Transition of high-grade cervicalintraepithelial neoplasia to micro-invasive carcinoma is characterized byintegration of HPV 16/18 and numerical chromosome abnormalities, J. Pathol., 202, 23 (2004). [ Links ]
18. A.P.Cullen, R. Reid y M. Campion, Analysis of the physical state of different humanpapillomavirus ADNs in intraepithelial and invasive cervical neoplasm, J.Virol., 65, 606 (1991). [ Links ]
19. L.Pirami, V. Giache, A. Becciolini, Analysis of VPH16, 18, 31, and 35 ADN inpre-invasive and invasive lesions of the uterine cervix, J. Clin. Pathol.,50, 600 (1997). [ Links ]
20. G. Hudelist, M. Manavi y K.I. Pischinger,Physical state and expression of HPV DNA in benign and dysplastic cervical tissue:different levels of viral integration are correlated with lesion grade, Gynecol.Oncol., 92, 873 (2004). [ Links ]
21. N.Sathish, P. Abraham y A. Peedicayil, Human papillomavirus 16 E6/E7 transcriptand E2 gene status in patients with cervical neoplasia, Mol. Diagn., 8,57 (2004). [ Links ]
22. K.B.Choo, C.C. Pan y M.S. Liu, Presence of episomal and integrated humanpapillomavirus DNA sequences in cervical carcinoma, J. Med. Virol., 21,101 (1987). [ Links ]
23. H.Lehn, L.L. Villa y F. Marziona, Physical state and biological activity of humanpapillomavirus genomes in precancerous lesions of the female genital tract, J.Gen. Virol., 69, 187 (1988). [ Links ]
24. J.Tidy, K.H. Vousden y P. Mason, A novel deletion within the upstream regulatoryregion of episomal human papillomavirus type 16, J. Gen. Virol., 70, 999(1989). [ Links ]
25. M.Fukushima, Y. Yamakawa y S. Shimano, The physical state of human papillomavirus16 DNA in cervical carcinoma and cervical intraepithelial neoplasia, Cancer.,66, 2155 (1990). [ Links ]
26. E.Kristiansen, A. Jenkins y R. Holm, Coexistence of episomal and integrated HPV16ADN in squamous cell carcinoma of the cervix, J. Clin. Pathol., 47, 253(1994). [ Links ]
27. J.Berumen, L. Casas y E. Segura, Genome amplification of human papillomavirustypes 16 and 18 incervical carcinomas is related to the retention of E1/E2 genes, Int. J.Cancer., 56, 640 (1994). [ Links ]
28. J.S.Park, E.S. Hwang y S.N. Park, Physical status and expression of VPH genes incervical cancers, Gynecol. Oncol., 65, 121 (1997). [ Links ]
29. S.D.Vernon, E.R. Unger y D.L. Miller, Association of human papillomavirus type 16integration in the E2 gene with poor disease-free survival from cervicalcancer, Int. J. Cancer., 74, 50 (1997). [ Links ]
30. M.Kalantari, F. Karlsen y G. Kristensen, Disruption of the E1 and E2 readingframes of VPH 16 incervical carcinoma is associated with poor prognosis, Int. J. Gynecol.Pathol., 17, 146 (1998). [ Links ]
31. R.Klaes, S.M. Woerner y R. Ridder, Detection of high-risk cervicalintraepithelial neoplasia and cervical cancer by amplification of transcriptsderived from integrated papillomavirus oncogenes, Cancer. Res., 59, 6132(1999). [ Links ]
32. M. Yoshinouchi, A. Hongo y K. Nakamura,Analysis by Multiplex PCR of the physical status of human papillomavirus type16 DNA in cervical cancers, J. Clin. Microbiol., 37, 3514 (1999). [ Links ]
33. M.Kalantari, E. Blennow y B. Hagmar, Physical state of HPV16 and chromosomalmapping of the integrated form in cervical carcinomas, Diagn. Mol. Pathol.,10, 46 (2001). [ Links ]
34. S.A.Tonon, M.A. Picconi y P.D. Bos, Physical status of the E2 human papilloma virus16 viral gene in cervical preneoplastic and neoplastic lesions, J. Clin.Virol. 21, 129 (2001). [ Links ]
35. P.Peitsaro, B. Johansson y S. Syrjanen, Integrated human papillomavirus type 16is frequently found in cervical cancer precursors as demonstrated by a novelquantitative real-time PCR technique, J. Clin. Microbiol., 40, 886(2002). [ Links ]
36. G.Badaracco, A. Venuti y A. Sedati, HPV16 and VPH18 in genital tumors:Significantly different levels of viral integration and correlation to tumorinvasiveness, J. Med. Virol., 67, 574 (2002). [ Links ]
37. S.Nagao, M. Yoshinouchi y Y. Miyagi, Rapid and sensitive detection of physicalstatus of human papillomavirus type 16 DNA by quantitative real-time PCR, J.Clin. Microbiol., 40, 863 (2002). [ Links ]
38. L.do Horto, V. Rodriguez y A.P. de Salles, HPV 16 detection in cervical lesions,physical state of viral DNA and changes in p53 gene, Sao Paulo Med. J., 121, 67 (2003). [ Links ]
39. G.Gallo, M. Bibbo y L. Bagella, Study of viral integration of HPV-16 in young patients withLSIL, J. Clin. Pathol., 56, 532 (2003). [ Links ]
40. W.Alazawi, M. Pett y S. Strauss, Genomic imbalances in 70 snap-frozen cervicalsquamous intraepithelial lesions: associations with lesion grade, state of theHPV16 E2 gene and clinical outcome, Br. J. Cancer., 91, 2063 (2004). [ Links ]
41. Z.Hernadi, T. Sapy y Z.T. Krasznai, The prevalence of the HPV 16 genome,integrated viral status and p53 genotype in cervical cancer population ofnorth-eastern Hungary,the correlation with the established markers of tumour progression, Eur. J.Obstet. Gynecol. Reprod. Biol., 13, 83 (2004). [ Links ]
42. S.Andersson, H. Safari y M. Mints, Type distribution, viral load and integrationstatus of high-risk human papillomaviruses in pre-stages of cervical cancer(CIN), Br. J. Cancer., 92, 2195 (2005). [ Links ]
43. A.H. Hopman, M.A. Kamps y F. Smedts, HPV insitu hybridization: Impact of different protocols on the detection ofintegrated HPV, Int. J. Cancer., 115, 419 (2005). [ Links ]
44. C.M.Ho, T.Y. Chien y S.H. Huang, Integrated human papillomavirus types 52 and 58are infrequently found in cervical cancer, and high viral loads predict risk ofcervical cancer, Gynecol. Oncol., Corrected Proof. (2005). [ Links ]
45. K.B.Choo, C.C. Pan y S.H. Han, Integration of human papillomavirus type 16 intocellular DNA of cervical carcinoma: preferential deletion of the E2 gene andinvariable retention of the long control region and the E6/E7 open readingframes, Virology, 161, 259 (1987). [ Links ]
46. C.M.Chen, M.P. Shyu y L.C. Au, Analysis of deletion of the integrated humanpapillomavirus 16 sequence in cervical cancer: a rapid multiplex polymerasechain reaction approach, J. Med. Virol. 44, 206 (1994). [ Links ]
47. S.A.Corden, L.J. Sant-Cassia y A.J. Easton, The integration of VPH-18 ADN incervical carcinoma, Mol. Pathol., 52, 275 (1999). [ Links ]
48. F.Luft, R. Klaes y M. Nees, Detection of integrated papillomavirus sequences byligation-mediated PCR (DIPS-PCR) and molecular characterization in cervicalcancer cells, Int. J. Cancer., 92, 9 (2001). [ Links ]
49. M.K.el Awady, J.B. Kaplan y S.J. O"Brien, Molecular analysis of integrated humanpapillomavirus 16 sequences in the cervical cancer cell line SiHa, Virology., 159, 389 (1987). [ Links ]
50. S.Schneider-Maunoury, O. Croissant y G. Orth, Integration of human papillomavirustype 16 DNA sequences: a possible early event in the progression of genitaltumors, J. Virol., 61, 3295 (1987). [ Links ]
51. K.B.Choo, H.H. Lee y C.C. Pan, Sequence duplication and internal deletion in theintegrated human papillomavirus type 16 genome cloned from a cervicalcarcinoma, J. Virol., 62,1659 (1988). [ Links ]
52. M.Wagatsuma, K. Hashimoto y T. Matsukura, Analysis of integrated humanpapillomavirus type 16 ADN in cervical cancers: amplification of viralsequences together with cellular flanking sequences, J. Virol., 64, 813(1990). [ Links ]
53. H.Lewis, K. Webster y A.M Sanchez-Perez, Cellular transcription factors regulatehuman papillomavirus type 16 gene expression by binding to a subset of the ADNsequences recognized by the viral E2 protein, J. Gen. Virol., 80, 2087(1999). [ Links ]
54. M.R Pett, W.O. Alazawi e I. Roberts,Acquisition of high-level chromosomal instability is associated withintegration of human papillomavirus type 16 in cervical keratinocytes, Cancer. Res., 64, 1359 (2004). [ Links ]
55. J.J. Dowhanick, A.A. McBride y P.M.Howley, Suppression of cellular proliferation by the papillomavirus E2 protein, J. Virol., 69, 7791 (1995). [ Links ]
56. E.C. Goodwin y D. DiMaio, Repression ofhuman papillomavirus oncogenes in HeLa cervical carcinoma cells causes theorderly reactivation of dormant tumor suppressor pathways, Proc. Natl. Acad.Sci. USA., 97, 12513 (2000). [ Links ]
57. S.L. Wells, D.A. Francis y A.Y. Karpova,Papillomavirus E2 induces senescence in HPV-positive cells via pRB and p21(CIP) dependent pathways, EMBO J., 19, 62 (2000). [ Links ]
58. D.A. Francis, S.I. Schmid y P.M. Howley,Repression of the integrated papillomavirus E6/E7 promoter is required forgrowth suppression of cervical cancer cells, J. Virol., 74, 2679 (2000). [ Links ]
59. A.M. Sanchez-Perez, S. Soriano y A.R.Clarke, Disruption of the human papillomavirus type 16 E2 gene protectscervical carcinoma cells from E2F-induced apoptosis, J. Gen. Virol., 78,3009 (1997). [ Links ]
60. S. Blachon, S. Bellanger y C. Demeret,Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteinsinduces apoptosis, J. Biol. Chem., 280, 36088 (2005). [ Links ]
61. H. Romanczuk y P.M. Howley, Disruption ofeither the E1 or E2 regulatory gene of human papillomavirus type 16 increaeseviral immortalization capacity, Proc. Natl. Acad. Sci. USA., 9, 3159(1992). [ Links ]
62. S. Jeon y P.F. Lambert, Integration ofhuman papillomavirus type 16 DNA into the human genome leads to increasedstability of E6 and E7 mRNAs: implications for cervical carcinogenesis, Proc.Natl. Acad. Sci. USA., 92, 1654 (1995). [ Links ]
63. W. Alazawi, M. Pett y B. Arch, Changes incervical keratinocyte gene expression associated with integration of humanpapillomavirus 16BFB, Cancer. Res., 62, 6959 (2002). [ Links ]
64. Jeon S, Allen-Hoffmann BL, Lambert PF,Integration of human papillomavirus type 16 into the human genome correlateswith a selective growth advantage of cells, J. Virol., 69, 2989 (1995). [ Links ]
65. K. Lindel, P. Burri y H.U. Studer, Humanpapillomavirus status in advanced cervical cancer: predictive and prognosticsignificance for curative radiation treatment, Int. J. Gynecol. Cancer.,15, 278 (2005). [ Links ]
66. B.A.Van Tine, J. Knops y T.R. Broker, Insitu analysis of the transcriptional activity of integrated viral DNA usingtyramide-FISH, Dev. Biol., 106, 381 (2001). [ Links ]
67. M.S. Rajeevan, D.C. Swan y R. Nisenbaum,Epidemiologic and viral factors associated with cervical neoplasia in HPV-16-positivewomen, Int. J. Cancer., 115, 114 (2005). [ Links ]
68. M. Dürst, C.M. Croce y CM, Gissmann,Papillomavirus sequences integrate near cellular oncogenes in some cervicalcarcinomas, Proc. Natl. Acad. Sci. USA., 84, 1070 (1987). [ Links ]
69. A. Mincheva, L. Gissmann y L. Zur Hausen,Chromosomal integration sites of human papillomavirus DNA in three cervicalcancer cell lines mapped by in situ hybridization, Med. Microbiol.Immunol., 176, 245 (1987). [ Links ]
70. L.A. Cannizzaro, M. Durst y M.J. Mendez,Regional chromosome localization of human papillomavirus integration sites nearfragile sites, oncogenes, and cancer chromosome breakpoints, Cancer. Genet.Cytogenet., 33, 93 (1988). [ Links ]
71. A. Parton, R.J. Grand y P. Biggs,Integrated HPV 1 genomas in a human keratinocyte cell line can betransactivated by a SV40/BPV1 recombinant virus which expresses BPV1 E2proteins, Virology., 175, 508 (1990). [ Links ]
72. J. Couturier, X. Sastre-Garau y S.Schneider-Maunoury, Integration of papillomavirus DNA near myc genes in genitalcarcinomas and its consequences for proto-oncogene expression, J. Virol., 65,4534 (1991). [ Links ]
73. T. Hori, H. Ichimura y Y. Minamihisamatsu,Chromosomal insertion and amplification of human papillomavirus 16 DNAsequences in a cell line of argyrophil small cell carcinoma of the uterinecervix, Jpn. J. Cancer. Res., 82, 371 (1991). [ Links ]
74. P.P. Smith, C.L. Friedman y E.M. Bryant,Viral integration and fragile sites in human papillomavirus-immortalized humankeratinocyte cell lines, Gen. Chrom. Cancer, 5, 150 (1992). [ Links ]
75. T. Kahn, E. Turazza y R. Ojeda,Integration of human papillomavirus type 6a DNA in a tonsillar carcinoma:chromosomal localization and nucleotide sequence of the genomic target region, Cancer.Res., 64, 1305 (1994). [ Links ]
76. C. Gilles, J. Piette y D. Ploton, Viralintegration sites in human papilloma virus-33-immortalized cervicalkeratinocyte cell lines, Cancer. Genet. Cytogenet., 90, 63 (1996). [ Links ]
77. K.B.Choo, C.M. Chen y C.P. Han, Molecular analysis of cellular loci disrupted bypapillomavirus 16 integration in cervical cancer: frequent viral integration intopologically destabilized and transcriptionally active chromosomal regions,J. Med. Virol., 49,15 (1996). [ Links ]
78. S.Reuter, M. Bartelmann y M. Vogt, APM-1, a novel human gene, identified by aberrantco-transcription with papillomavirus oncogenes in a cervical carcinoma cellline, encodes a BTB/POZ-zinc finger protein with growth inhibitory activity, EMBOJ., 17, 215 (1998). [ Links ]
79. L.A.Koopman, K. Szuhai K y J.D. Van Eendenburg, Recurrent integration of humanpapillomaviruses 16, 45, and 67 near translocation breakpoints in new cervicalcancer cell lines, Cancer. Res., 59, 5615 (1999). [ Links ]
80. K.A.Shera, C.A. Shera y J.K. McDougall, Small tumor virus genomes are integratednear nuclear matrix attachment regions in transformed cells, J. Virol., 75, 12339 (2001). [ Links ]
81. N.Wentzensen, R. Ridder y R. Klaes, Characterization of viral-cellular fusiontranscripts in a large series of HPV16 and 18 positive anogenital lesions, Oncogene., 21, 419 (2002). [ Links ]
82. T.Wiest, E. Schwarz y C. Enders, Involvement of intact HPV16 E6/E7 geneexpression in head and neck cancers with unaltered p53 status and perturbed pRbcell cycle control, Oncogene., 21, 1510 (2002). [ Links ]
83. E.C.Thorland, S.L. Myers y B.S. Gostout, Common fragile sites arepreferential targets for HPV16 integrations in cervical tumors, Oncogene., 22, 1225 (2003). [ Links ]
84. M.JFerber, D.P. Montoya y C. Yu, Integrations of the hepatitis B virus (HBV) andhuman papillomavirus (HPV) into the human telomerase reverse transcriptase(hTERT) gene in liver and cervical cancers, Oncogene., 22, 3813 (2003). [ Links ]
85. M.J.Ferber, E.C. Thorland y A.A. Brink, Preferential integration of humanpapillomavirus type 18 near the c-myc locus incervical carcinoma, Oncogene., 22, 7233 (2003). [ Links ]
86. T.Yu, M.J. Ferber y T.H. Cheung, The role of viral integration in the developmentof cervical cancer, Cancer. Genet. Cytogenet., 158, 27 (2005). [ Links ]
87. E. Klimov, S. Vinokourova y E. Moisjak,Human papilloma viruses and cervical tumours: mapping of integration sites andanalysis of adjacent cellular sequences, BMC Cancer, 2, 24 (2002). [ Links ]
88. P.Peitsaro, M. Ruutu y S. Syrjanen, Divergent expression changes of telomeraseand E6/E7 mRNA, following integration of human papillomavirus type 33 in cultured epithelialcells, Scand. J. Infect. Dis., 36, 302 (2004). [ Links ]
89. A.J. Klingelhutz, S.A. Foster y J.K. McDougall,Telomerase activation by the E6 gene product of human papillomavirus type 16, Nature,380 , 79 (1996). [ Links ]
90. A.Hidalgo, C. Schewe y S. Petersen, Human papilloma virus status and chromosomalimbalances in primary cervical carcinomas and tumour cell lines, Eur. J.Cancer, 36, 542 (2000). [ Links ]
91. A.Hidalgo, A. Monroy y R. Arana, Chromosomal imbalances in four new uterinecervix carcinoma derived cell lines, BMC Cancer, 3, 8 (2003). [ Links ]
92. L.Casas, S.C. Galván y R.M. Ordóñez, Asian-American variants of humanpapillomavirus type 16 have extensive mutations in the E2 gene and are highlyamplified in cervical carcinomas, Int. J. Cancer., 83, 449 (1999). [ Links ]
93. R.M.Ordóñez, A.M. Espinosa y D.J. Sanchez-Gonzalez, Enhanced oncogenicity ofAsian-American human papillomavirus 16 is associated with impaired E2repression of E6/E7 oncogene transcription, J. Gen. Virol., 6, 1433(2004). [ Links ]
94. G.Veress, K. Szrka y X.-P. Dong, Functional significance of sequence variation inthe E2 gene and the long control region of human papillomavirus type 16, J.Gen. Virol., 80, 1035 (1999). [ Links ]
95. T.Turan, M. Kalantari. I.E. Calleja Macias, Methylation of the humanpapillomavirus 18 L1 gene: a biomarker of neoplastic progression, Virol., 349,175 (2006). [ Links ]
96. M.Kalantari, I.E Calleja-Macias y D. Tewari, Conserved methylation patterns ofhuman papillomavirus type 16 DNA in asymptomatic infection and cervicalneoplasia, J. Virol., 78,12762 (2004). [ Links ]
97. S.Badal, V. Badal E I.E. Calleja-Macias, The human papillomavirus-18 genome isefficiently targeted by cellular DNA methylation, Virol., 324,483(2004). [ Links ]
98. V. Badal, L.S. Chuang y E.H. Tan, CpGmethylation of human papillomavirus type 16 DNA in cervical cancer cell linesand in clinical specimens: genomic hypomethylation correlates with carcinogenicprogression, J. Virol., 77, 6227 (2003). [ Links ]
99. M. van Duin, P.J.F. Snijders y H.F.J.Schijnemakers, Human papillomavirus 16 load in normal and abnormal cervicalscrapes: an indicator of NICII/III and viral clearance, Int. J. Cancer., 98, 590 (2002). [ Links ]
100. N. Schlecht, A. Trevisan y E.Duarte-Franco, Viral load as a predictor of the risk of cervicalintraephitelial neoplasia, Int. J. Cancer, 103, 519 (2003). [ Links ]
101. V. Dalstein, D. Riethmuller y J.L. Pretet,Persistence and load of high-risk HPV are predictors for development ofhigh-grade cervical lesions: a longitudinal French cohort study, Int. J.Cancer., 106, 396 (2003). [ Links ]
102. B.C. Das, J.K. Sharma y V. Gopalakrishna,Analysis by polymerase chain reaction of the physical state of humanpapillomavirus type 16 DNA in cervical preneoplastic and neoplasic lesion, J.Gen. Virol., 73, 2327 (1992). [ Links ]
103. K. Cooper y C.S. Herrintong, Stickland JE,Episomal and integrated human papillomavirus in cervical neoplasia shown bynon-isotopic in situ hybridization, J. Clin. Pathol., 44, 990(1991). [ Links ]
104. A. Venuti y M.L. Marcante, Presence ofHuman papillomavirus type 18 DNA in vulvar carcinomas and its integration intothe cell genome, J. Gen. Virol., 70, 1587 (1989). [ Links ]
105. T. Fuji, N.Masumoto y M. Saito, Comparison between in situ hybridization andreal-time PCR technique as a means of detecting the integrated form of humanpapillomavirus 16 incervical neoplasia, Diagn. Mol. Pathol., 14, 103 (2005). [ Links ]
106. M.F.Evans y K. Cooper, Human papillomavirus integration: detection by in situ hybridization and potential clinical application, J. Pathol., 202, 1(2004). [ Links ]

