










Services on Demand
Journal
Article
 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
Print version ISSN 0034-7418On-line version ISSN 1909-6356
Rev. colomb. cienc. quim. farm. vol.35 no.2 Bogotá July/Dec. 2006
Artículo de investigación
Desenvolvimento e validação de um método analítico para quantificação por espectroscopia UV de captopril em comprimidos de liberação prolongada
Development and validation of an UV analytic method intended for quantification of captopril in delayed release tablets
Hellen Karine Stulzer1, Monika Piazzon Tagliari1, Marcos Antonio Segatto Silva1
1 Laboratório de Controle de Qualidade, Departamento de Ciências Farmacêuticas , Centro de Ciências da Saúde, Universidade Federal de Santa Catarina, Campus Trindade, 88040-900, Florianópolis, SC, Brasil.
E-mail: hellen.stulzer@gmail.com
Recibido para evaluación: octubre 5 de 2006 Aceptado para publicación: diciembre 18 de 2006
RESUMO
Um método analítico utilizando espectroscopia no ultravioleta (UV) foi desenvolvido e validado para quantificar o fármaco captopril em comprimidos de liberação prolongada. Os parâmetros utilizados no processo de validação foram especificidade, linearidade e intervalo, precisão, exatidão e robustez. A linearidade no intervalo de 5.0 - 40.0 µg/mL apresentou um coeficiente de correlação de 0,9998. Os excipientes das formulações não interferiram com a análise e a recuperação da amostra foi de 100,20 ± 0,28 %. Todos os resultados foram satisfatórios e o método provou ser adequado para quantificar o captopril nos comprimidos de liberação prolongada.
Palavras chave: captopril, validação analítica, espectroscopia UV
SUMMARY
An analytical method using an ultraviolet spectroscopy (UV) was developed and validated to quantify the drug captopril in tablets of delayed release. The available parameters were: specificity, linearity and range, precision, accuracy and robustness. The linearity in the range of 5.0 - 40.0 µg/mL presented a correlation coefficient of 0, 9998. The excipients in the formulation did not interfere with the analysis and the recovery was quantitative 100, 20 ± 0, 28%. All results were satisfactory and the method proved to be adequate to quantify captopril in delayed release tablets.
Key words: Captopril, Analytical Validation, UV Spectroscopy
INTRODUÇÃO
O captopril, que corresponde ao D-2-metil-3-mercaptopropanol-L-prolina (figura 1) é um agente anti-hipertensivo ativo oralmente e atua através da inibição competitiva da enzima conversora de angiotensina. Este fármaco demonstra excelente efetividade no tratamento da hipertensão arterial, sendo considerado o medicamento de primeira escolha. Porém, o seu tempo de ação é limitado, apenas de 6 a 8 horas, e necessita ser administrado três vezes ao dia. Por tanto, formulações de liberação prolongada desse fármaco vem sendo alvo de constantes pesquisas no intuito de diminuir a freqüência de administração do medicamento, facilitando a adesão ao tratamento (1-6).

Na literatura são relatados diversos métodos para quantificação do captopril matéria prima e em formulações farmacêuticas convencionais, como cromatografia líquida de alta eficiência (CLAE), volumetria, métodos fluorométricos e colorimétricos, espectroscopia Raman, eletroforese e espectroscopia no ultravioleta. Segundo a United States Pharmacopeia 26, o captopril matéria prima pode ser quantificado por titulação e CLAE, já para os comprimidos de liberação imediata. Além destas técnicas, a espectroscopia no ultravioleta também é indicada para quantificação do captopril durante o ensaio de dissolução (7-13).
Diante desses dados, o presente trabalho propõe o desenvolvimento e a validação de um método analítico que atenda as necessidades para quantificação do captopril matéria prima e o contido em comprimidos de liberação prolongada, pela técnica de espectroscopia de absorção no ultravioleta (UV).MATERIAIS E MÉTODOS
Materiais
Captopril ([2S-1-(3-mercapto-2-methylpropioniyl)-L-proline]) é proveniente da Shenyang Fine Chemical Co., China. O padrão de captopril utilizado foi grau USP com pureza de 99,99%. Os excipientes utilizados nos grânulos e comprimidos foram celulose microcristalina 101 e 102, etilcelulose (Ethocel 10STD, Colorcon, USA), metilcelulose (Methocel E 15 PR, Colorcon, USA), lactose monohidrata, lactose Supertab®, ácido esteárico, estearato de magnésio, dióxido de silício coloidal (Aerosil®) e polivinilpirrolidone (PVP K30). Todos os reagentes empregados apresentavam grau analítico.
Métodos
Desenvolvimento dos comprimidos
Os comprimidos de 45 mg de captopril (tabela 1) foram produzidos a partir de grânulos revestidos em leito fluidizado (NARONG modelo FBS-1) com diferentes polímeros. Os grânulos revestidos foram misturados em minimisturador marca LAWES durante 15 minutos, junto com Aerosil®, ácido esteárico, celulose microcristalina 102 e lactose Supertab®. Após a mistura foi efetuada a compressão direta em compressora LAWES modelo 2000 10 PSO, com punção bicôncavo de 7 mm.
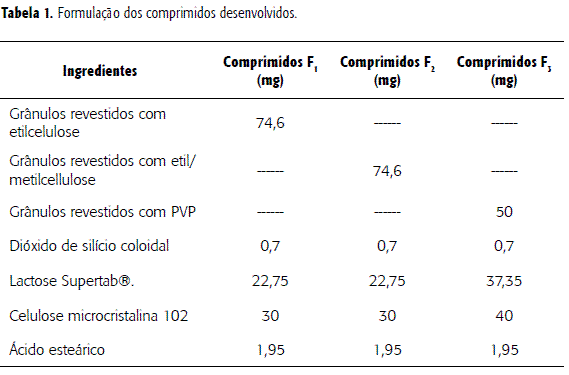
Avaliação do espectrofotômetro UV
A calibração do espectrofotômetro (BECKMAN COULTER, modelo DU 640) foi realizada conforme a British Pharmacopoeia (14). Foram avaliados o controle das absorbâncias, limite de luz parasita e resolução.
Validação analítica
1. Especificidade
Nos estudos de especificidade de métodos para determinação do teor de fármaco, procedeu-se analisando a solução padrão de captopril e os excipientes utilizados isoladamente, dissolvidos em tampão fosfato de sódio 0,05 N pH 7,0 através de uma varredura entre 200 e 400 nm em espectrofotômetro UV.
2. Linearidade e intervalo
A linearidade e o intervalo foram determinados com a construção da curva de calibração (em triplicata), obtida através das leituras da absorbância em 212 nm de diferentes concentrações da amostra (5 a 40 μg/mL em tampão fosfato de sódio 0,05 N pH 7,0), seguindo a lei de Lambert-Beer que preconiza que a reta traçada deve passar por todos os pontos do intervalo (15).
3. Exatidão
A exatidão do método foi determinada através do ensaio de recuperação, onde diferentes concentrações de captopril (15, 30 e 45 mg) foram adicionadas à mistura de excipientes que compõe a formulação. Estes foram pesados em balança analítica e devidamente homogeneizados em gral de porcelana durante 15 minutos. Em seguida pesou-se quantidades da mistura correspondentes a 15, 30 e 45 mg, transferindo-as para um balão volumétrico de 100 mL, onde o volume foi completado com tampão fosfato de sódio 0,05 N pH 7,0. Estas soluções permaneceram em banho de ultra-som durante 15 minutos, sendo em seguida centrifugadas durante 10 minutos à 4000 rpm. Destes balões foram feitas diluições a fim de que a concentração final de captopril abrangesse a linearidade do método.
4. Precisão
Preparo da amostra
10 comprimidos de liberação prolongada de captopril (45 mg/comprimido) foram triturados em gral de porcelana. Em seguida pesou-se analiticamente o equivalente a média das massas dos 10 comprimidos e transferiu-se para um balão volumétrico de 100 mL, onde o volume foi completado com tampão fosfato de sódio 0,05 N pH 7,0. Esta solução permaneceu em banho de ultra-som durante 15 minutos, sendo em seguida centrifugada durante 10 minutos à 4000 rpm. Alíquota equivalente a 2 mL do sobrenadante foi adicionada em balão volumétrico de 50 mL e o volume completado com tampão fosfato de sódio 0,05 N pH 7,0.
Este parâmetro foi avaliado em dois níveis:
-
Repetibilidade (precisão intracorrida)
A repetibilidade do método foi determinada através de 3 repetições, contendo seis replicatas a 100% da concentração do teste (conforme descrito no item preparo da amostra), realizadas pelo mesmo analista, no mesmo laboratório, em períodos alternados (manhã, tarde e noite), verificando se os resultados encontram-se dentro da máxima diferença aceitável.
-
Precisão intermediária
A precisão intermediária foi expressa através de variações dentro do mesmo laboratório, em dias diferentes por analistas diferentes. O ensaio foi realizado com 6 replicatas a 100% da concentração do teste (conforme descrito no item preparo da amostra), com 3 analistas diferentes.
5. Robustez
A robustez do método foi determinada por análises das amostras (18 µg/mL) sob pequenas variações do pH 7,0 ± 0,5 do tampão fosfato
Análises estatísticas
A análise estatística dos dados foi realizada através de análise de variância ANOVA unifatorial, onde os resultados são considerados significativos quando a probabilidade é inferior a 5% (p < 0,05/ intervalo de confiança de 95%) , teste t de Student com nível de significância α = 0,05 (intervalo de confiança de 95%) e teste de Cochran. A avaliação estatística dos resultados foi realizada através do Software ORIGIN 70 e MS Excel®.
RESULTADOS E DISCUSSÕES
Avaliação do espectrofotômetro
Como o espectrofotômetro foi o equipamento utilizado no desenvolvimento do método de quantificação do captopril, foi submetido a uma série de análises, objetivando avaliar a confiabilidade dos resultados obtidos com este equipamento, de acordo com a British Pharmacopoeia (14).
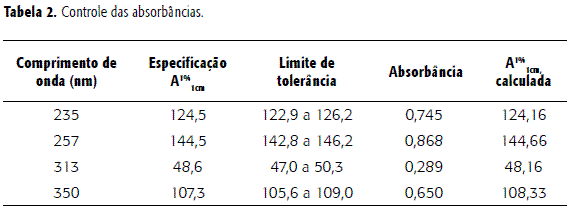
Controle das absorbâncias
Para avaliar a intensidade de emissão dos comprimentos de onda da faixa UV, foi realizado o ensaio com solução de dicromato de potássio 0,006% em H2SO4 0,005 M, medindo-se a absorbância e calculando-se a absortividade específica (A1%1cm) em comprimentos de onda pré-estabelecidos (14). Os resultados alcançados, descritos na Tabela 2, estão de acordo com os valores preconizados pelo limite de tolerância.
Luz parasita
A solução de dicromato de potássio 1,2 % m/v, medida em uma cubeta de quartzo de 1 cm em 220 nm, apresentou absorbância de 1,87 u.a, estando de acordo com o preconizado, que estabelece valor máximo de 2 u.a (14).
Resolução
A resolução do aparelho no caso de análise qualitativa preconiza a realização de uma varredura de uma solução de tolueno em hexano (0,02 % v/v). A solução deve apresentar valor máximo de absorbância em 269 nm e mínimo em 266 nm. As varreduras apresentaram-se de acordo com o preconizado (14).
Validação analítica
A United States Pharmacopeia 26 (13) e a International Conference on Harmonization Q-2B (16) preconizam para a validação de métodos de doseamento que sejam realizados os ensaios de exatidão, precisão, especificidade, linearidade e intervalo. Neste trabalho, além destes parâmetros especificados, a robustez também foi analisada.
Especificidade
Este parâmetro é expresso como a capacidade que o método possui de medir exatamente um composto em presença de outros componentes, como impurezas, produtos de degradação e componentes da matriz (13,16,17). Porém, neste ensaio foi avaliada somente a influência dos componentes da matriz. Os resultados para especificidade demonstraram que o método é adequado em relação ao parâmetro avaliado, pois não ocorreu pico de absorção dos excipientes na faixa de luz próximo a 212 nm (Figura 2), comprimento de onda de absorção máxima para o captopril.
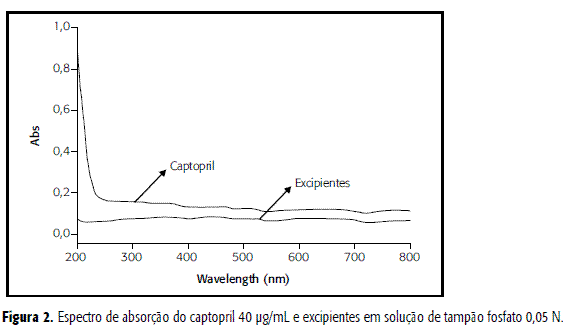
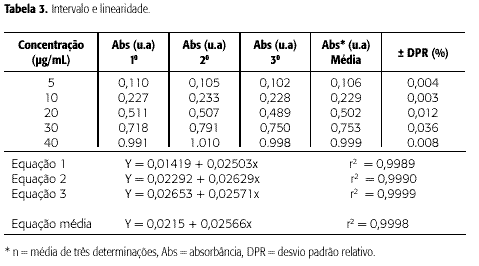
Linearidade e intervalo
A linearidade de uma metodologia analítica é a capacidade de demonstrar que os resultados obtidos são proporcionais à concentração do analito, num intervalo especificado. Sendo este a faixa entre os limites de quantificação superior e inferior de um método analítico (13,16).
Na Tabela 3 e Figura 3, observa-se que o método apresentou linearidade no intervalo da concentração testada (5 a 40 μg/mL). Esta faixa de concentração possibilitou detectar o captopril dentro dos limites de interesse e a resposta do detector de ultravioleta conservou-se linear. Através da análise de variância ANOVA e teste t Student observou-se que o valor do intercepto não é estatisticamente diferente de zero e os desvios da linearidade são estatisticamente iguais; o valor de F encontrado foi de 0,0035 com F de significação igual a 3,88. O desvio padrão encontrado para os diferentes valores de A (coeficiente angular) na equação da reta foi de 0,06, indicando que as mesmas podem ser sobrepostas. Para o intercepto e coeficiente de correlação, os desvios padrões encontrados foram de 3,6 e 3,1, respectivamente.

Exatidão
A exatidão dum método analítico é a proximidade dos resultados obtidos pelo método com o valor verdadeiro. Nos casos em que amostras de todos os componentes do medicamento estão disponíveis, se aceita a análise pelo método de adição do padrão, no qual se adicionam quantidades conhecidas do padrão de referência aos excipientes da formulação (13,17). A tabela 4 apresenta os valores encontrados para recuperação do captopril nas diferentes concentrações.
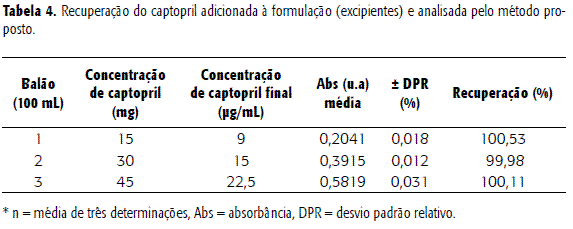
A porcentagem média para o ensaio de recuperação foi de 100, 20 ± 0,28 %, resultado que traduz a concordância com o valor verdadeiro da amostra. Através do teste t de Student, pode-se afirmar que o valor de recuperação encontrado é estatisticamente igual a 100% para nível de significância α = 0,05. Através do teste de Cochran, que avalia a diferença de variações em função da porcentagem de adição, pode-se verificar que a adição de diferentes concentrações não influenciou nas variabilidades dos resultados.
Precisão
A precisão é a avaliação da proximidade dos resultados obtidos em uma série de medidas de uma amostragem múltipla de uma mesma amostra (13,17). Este parâmetro foi avaliado quanto aos níveis de repetibilidade e a precisão intermediária, conforme tabelas 5 e 6.
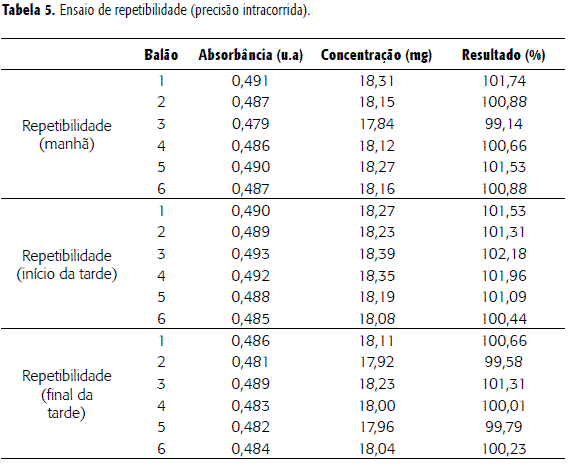
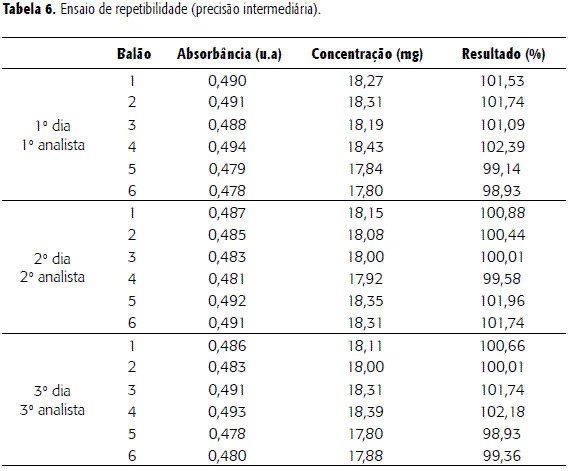
Neste ensaio foram utilizadas seis determinações onde observamos a diferença entre as repetições. O ensaio apresentou um desvio padrão relativo (DPR) de 0,8452%; está este valor dentro da variação aceitável onde o limite máximo de DPR seria ± 2,0 %. A análise de ANOVA demonstrou que não há diferença estatística significativa entre as análises dos diferentes períodos; o valor de F calculado foi 1,088 e F de significação igual a 3,68 e p = 0,36.
Para a repetibilidade, o valor da média encontrado foi igual a 100,68%; o desvio padrão relativo foi 1,17% estando dentro da faixa de aceitabilidade que estabelece valores de DPR ± 2,0 %. A análise de variância ANOVA demonstrou que o valor de F encontrado foi de 0,122451 com F de significação igual a 3,68232 e p = 0,88. O teste Student indicou que não há diferença significativa entre os grupos. Assim, pode-se observar que dias diferentes bem como analistas diferentes não promoveram diferenças significativas nos resultados das análises.
Robustez
A robustez dum método analítico é a medida de sua capacidade em resistir a pequenas e deliberadas variações dos parâmetros analíticos (13, 17). Variações do pH do tampão fosfato de sódio 0,05 N pH 7,0 ± 0,5 não apresentaram efeitos significativos nos valores das absorbâncias. Os valores do desvio padrão relativo (DPR) encontrado foram igual a ± 0,657 e ± 0, 453 para os valores de pH de 6,95 e 7,5, respectivamente. Assim, o método se demonstrou robusto em relação a variações de pH.
CONCLUSÕES
A metodologia proposta para a determinação do captopril em comprimidos de liberação prolongada demonstrou ser um método simples, rápido e barato, atendendo as exigências quanto à especificidade, linearidade e intervalo, precisão, exatidão e robustez estes aspectos essenciais para a validação de metodologias analíticas. Os modelos estatísticos empregados nos dados dos diferentes parâmetros avaliados permitiram uma maior confiabilidade nos resultados. Dessa forma, os resultados foram considerados satisfatórios e o método provou ser adequado para quantificar o captopril nos comprimidos de liberação prolongada desenvolvidos.
AGRADECIMIENTOS
Os autores agradecem o apoio financeiro da Capes, Brasil, bem como a colaboração da Universidade Federal de Santa Catarina e Universidade do Vale do Itajaí.
REFERÊNCIAS
1. M.E. Franklin, P.V. Addison, W.D. Baker, Improved analytical procedure for the measurement of captopril in human plasma by gas chromatography-mass spectrometry and its application to pharmacokinetic studies, J. Chromatogr. B, 705, 47 (1998). [ Links ]
2. I.I. Salem, W.A. Saif, Y. Jmeian, A selective and rapid method for the quantification of captopril in human plasma using liquid chromatography/selected reaction monitoring mass spectrometry, J. Pharm. Biomed. Anal., 37, 1073 (2005). [ Links ]
3. J.J. Kang, I. Toma, A. Sipos, F. McCulloch, J. Peti-Peterdi, Imaging the renin angiotensin system: An important target of anti-hypertensive therapy, Adv. Drug. Delivery Rev., 58, 824 (2006). [ Links ]
4. H. Ho, H. Wang, M. Sheu, The evaluation of granulated excipients as matrix material for controlled delivery of captopril, J. Controlled Release, 49, 243 (1997). [ Links ]
5. A.O Nur, J.S. Zhang, Captopril floating and/or bioadhesive tablets: design and release kinetics, Drug. Dev. Ind. Pharm, 26, 965, (2000). [ Links ]
6. H.P. Rang, M.M. Dale, J.M. Ritter, Farmacologia. Guanabara Koogan Eds, Rio de Janeiro, 1999, p. 703. [ Links ]
7. S.A. Shama, A.E. Amin, H. Omara, Colorimetric microdetermination of captopril in pure form and in pharmaceutical formulations,J. Quant. Spectrosc. Radiat. Transfer, 102, 261 (2006). [ Links ]
8. S. Mazurek, R. Szostak, Quantitative determination of captopril and prednisolone in tablets by FT-Raman spectroscopy, J. Pharm. Biomed. Anal, 40, 1225, (2006). [ Links ]
9. N. Rahman, M. Singh, N. Hoda, Validation of simultaneous volumetric and spectrophotometric methods for the determination of captopril in pharmaceutical formulations,Il Farmaco, 60, 569 (2005). [ Links ]
10. A. El Gindy, S. Emara, G.M. Hadad, Determination of certain drugs in binary mixtures formulations by second derivative ratio spectrophotometry and LC,Il Farmaco, 59, 703 (2004). [ Links ]
11. M. Al-Ghannam, A.M. El-Brashy, B.S. Al-Farhan, Fluorimetric determination of some thiol compounds in their dosage forms, Il Farmaco, 57, 625 (2002). [ Links ]
12. I. Panderi, M.P. Parissi, Determination of captopril and captopril-hydrochlorothiazide combination in tablets by derivative UV spectrophotometry,Int. J. Pharm, 86, 99 (1992). [ Links ]
13. The United States Pharmacopeia, 26th ed., Rockville: United States Pharmacopeial Convention, 2003. [ Links ]
14. British Pharmacopoeia, London: Her Majesty´s Stationary Office, 2000. [ Links ]
15. A.I. Vogel, Análise química quantitativa, LTC, Eds, Rio de Janeiro, 1992, p. 713. [ Links ]
16. ICH - International Conference on Harmonization of Technical Requirements for registration of Pharmaceutical for Human use: Q 2B- validation of Analytical procedure: methodology, 1996. [ Links ]
17. European Pharmacopoeia, 4th ed., Strasbourg: Convention on the Elaboration of a European Pharmacopoeia, 2002. [ Links ]

