










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
Print version ISSN 0034-7418On-line version ISSN 1909-6356
Rev. colomb. cienc. quim. farm. vol.36 no.1 Bogotá Jan./June 2007
Artículo de investigación
Estandarización y validación de una metodología analítica para la determinación de carbamazepina en plasma mediante extracción en fase sólida y cromatografía líquida de alta eficiencia
Development and validation of an analytical methodology for the assay of carbamazepine in human plasma by solid phase extraction and high performance liquid chromatography
Jaime H. Rojas1, Noralba Sierra2
1 Universidad Nacional de Colombia, Departamento de Farmacia, A.A. 14490, Bogotá, D.C., Colombia. jhrojasb@unal.edu.co
2 Universidad Nacional de Colombia, Departamento de Farmacia, A.A. 14490, Bogotá, D.C., Colombia.
RESUMEN
En este artículo se presentan los resultados del desarrollo y estandarización de un método para la determinación de carbamazepina en plasma, fármaco ampliamente utilizado como anticonvulsivante. El método consiste, básicamente, en la separación cromatográfica por HPLC en fase inversa utilizando una columna C18 y una fase móvil formada por una mezcla de metanol-acetonitrilo-agua y detección a 213 nm. Como estándar interno se utilizó lorazepam.
El método desarrollado presenta un intervalo lineal entre 125 nanogramos y 10 microgramos por mililitro, concentraciones adecuadas para la determinación de carbamazepina en plasma de pacientes tratados con este fármaco. Igualmente demostró ser preciso, exacto y selectivo frente a los principales metabolitos de la carbamazepina y a los componentes endógenos del plasma.
Así mismo, se estudió la estabilidad de la carbamazepina en la fase móvil, requisito para el almacenamiento de muestras y de estándares. La selectividad y especificidad del método se estudió frente a otros dos fármacos utilizados para el tratamiento de la epilepsia, fenobarbital y difenilhidantoína.
El método se aplicó satisfactoriamente para la determinación de carbamazepina en muestras de plasma, y puede ser empleado para estudios farmacocinéticos, de biodisponibilidad y de bioequivalencia del fármaco, objetivos del estudio. Sus resultados también se pueden utilizar para establecer regímenes terapéutico.
Palabras clave: Carbamazepina (CBZ), lorazepam (LRZ), extracción en fase sólida, cromatografía líquida, octadecilsilano, detección espectrofotométrica, arreglo de diodos, validación.
SUMMARY
This paper describes the analytical results during validation of a reverse high performance liquid chromatography methodology for the assay of carbamazepine in human plasma, useful for their dosage in biodisponibility and bioequivalence studies. The stationary bounded phase used was octadecylsilane and the mobile phase was composed by a mixture of methanol-acetonitrile-water. The detection was performed by UV at 213 nm and lorazepam was utilized as internal standard. The method show linearity between 125 ng and 10 mcg/mL, amounts useful for the assay of carbamazepine in plasma.
Key words: Carbamazepine (CBZ), lorazepam (LRZ), solid phase extraction, high performance liquid chromatography, octadecylsilane, UV detection, diode array detector, validation.
INTRODUCCIÓN
La carbamazepina es un agente efectivo para controlar la epilepsia y tratar la neuralgia del trigémino (1), las enfermedades maníaco-depresivas y los pacientes que presenten crisis convulsivas generalizadas tónico-clónicas (2). Dentro de los varios metabolitos de la carbamazepina, el más importante desde el punto de vista tanto cualitativo como cuantitativo es el 10-11 epóxido de la carbamazepina, anticonvulsivante tan activo como la carbamazepina y que al igual que ésta se liga en alto porcentaje a las proteínas plasmáticas (2). Debido a su extendido uso, se debe disponer en lo posible de parámetros farmacocinéticas adecuados para establecer los diferentes regímenes terapéuticos, adecuados para el tratamiento de las distintas patologías.
La cromatografía líquida de alta eficiencia en fase inversa es, tal vez, la técnica más utilizada para la cuantificación de carbamazepina en fluidos biológicos (3). Saris y colaboradores (4) determinan CBZ y CBZ-diol en cabello por HPLC luego de un proceso de hidrólisis con NaOH y extracción en fase sólida. Mandrioli y colaboradores (5), con una fase móvil de acetonitrilo-metanol-buffer pH 1,9, reportaron CBZ y cuatro de sus metabolitos utilizando cartuchos Oasis-HLB y una columna C8 para la separación cromatográfica, y encontraron recuperaciones entre 84,8 y 103%. Este tipo de cartucho se utilizó también para determinar CBZ y otros fármacos en aguas residuales de hospitales por HPLC-MS (6). La detección se realiza generalmente al UV (3,4) y por otros procedimientos como fluorescencia inducida fotoquímicamente (7).
Igualmente se ha reportado la cromatografía en fase gaseosa para el análisis de CBZ y su metabolito 10-11 epóxido (8). Matthias y Wätzig (9) reportan el uso de cromatografía micelar electrocinética para controlar la pureza de CBZ a pH 9,2 con bórax y dodecilsulfato de sodio. Por otra parte, Raggi y colaboradores (10), mediante esta última técnica y empleo de un buffer de fosfatos pH 8 y metanol 95/5 separan, sobre capilares de sílica, CBZ de cinco de sus metabolitos; encuentran una concentración de 0,12 mcg/mL como límite de detección.
Para la validación se siguieron algunas de las pautas establecidas en la guía de la FDA-ICH para determinar las metodologías bioanalíticas (11).
PARTE EXPERIMENTAL
Equipos
Espectrofotómetro Unicam UV-VIS 2, cromatográfo Perkin Elmer con módulo para cuatro solventes, detector espectrofotométrico Waters 420, integrador Perkin Elmer LCI-100, baño de ultrasonido Erma T460/H, columna LiChrospher Merck RP 18 (125 x 4.6 mm, 5µm;, potenciómetro Methrom E-50, espectrofotómetro UV/VIS Unicam UV2, bomba de vacío Millipore, equipo de filtración Millipore columna C18 Lachrom.
Reactivos y materiales
Carbamazepina (CBZ) USP, nitrazepam, flunitrazepam, diazepam, lorazepam (LRZ), difenilhidantoína (DPH), fenobarbital (PNB), acetonitrilo HPLC, metanol HPLC, agua HPLC, nitrógeno, metanol RA, cartuchos Sep-Pak C18 para extracción en fase sólida, membranas Millipore 0.45 micrómetros HVLP, filtros para muestras Miles de Millipore.
Optimización del sistema cromatográfico
Como fases móviles se estudiaron mezclas de acetonitrilo-agua, metanol-acetonitrilo-agua y metanol-agua-trietilamina. Esta última mezcla se trabajó a tres valores diferentes de pH, 3.0, 5.0 y 7.0, empleando ácido fosfórico para ajustar los valores de pH.
Igualmente se trabajaron dos temperaturas y diferentes flujos de fase móvil, siempre con el propósito de mejorar la resolución del sistema cromatográfico.
Como moléculas susceptibles de emplear como estándar interno se estudiaron el lorazepam (LRZ), el flunitrazepam y el diazepam. Luego de algunos ensayos se seleccionó el LRZ como estándar interno. De acuerdo con los espectros de absorción obtenidos con CBZ y el LRZ, se estableció la longitud de onda para la detección cromatográfica (213 nm).
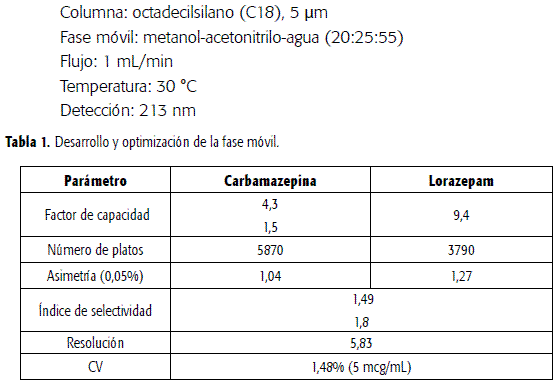
En el proceso de estandarización se realizaron modificaciones a la fase móvil y a la velocidad de flujo, utilizando como parámetros de evaluación el tiempo de retención y el factor de asimetría (Tabla 1), así como la selectividad entre los dos fármacos y con los componentes endógenos del plasma. Los parámetros calculados se encuentran dentro de lo establecido (11, 12).
Como condiciones finales del sistema cromatográfico se establecieron:
Estabilidad
Igualmente se estudió la estabilidad de CBZ en la fase móvil, requisito para el almacenamiento de muestras y de estándares. Una solución estándar de CBZ de concentración 5 mcg/mL en la fase móvil se guardó a -4 °C y se analizó cinco veces durante seis días, se observó un descenso significativo (prueba de t, α= 0,05) en el contenido al sexto día, indicando que los estándares se pueden conservar hasta por cinco días a -4 °C. Tratándose de muestras de plasma del voluntario, éstas se almacenaron a -30 °C.
Extracción en fase sólida
Para optimizar el proceso de extracción en fase sólida, se estudiaron soluciones estándar de CBZ y de LRZ, alícuotas de las cuales se depositaron sobre cartuchos de extracción previamente activados, para proceder luego a un lavado del cartucho seguido de la elusión y cuantificación de las dos sustancias. El lavado quedó determinado mediante la aplicación sucesiva de 2 y 1 mL agua, 1 mL de metanol al 20% y 100 µL de metanol. Como solventes de elusión se ensayaron metanol y metanol-etanol 70/30, obteniéndose con éste último las recuperaciones más altas. Para el lavado se pasó sucesivamente de 1 a 0,5 mL de la mezcla de metanol-etanol. El eluato se evaporó bajo atmósfera de nitrógeno a 40 °C, luego de lo cual se redisolvió el residuo en 0,5 mL de fase móvil y se filtró por membrana de 0,45 µm antes de la inyección. El procedimiento utilizado para las muestras fue similar.
RESULTADOS Y DISCUSIÓN
Selectividad
La selectividad del sistema cromatográfico se verificó frente a la fase móvil, a los componentes del plasma (Figura 1) y frente al estándar interno, LRZ (Figura 2). En la Figura 1, la flecha indica la posición o tiempo de retención de la CBZ.


Igualmente se observó la resolución frente al fenobarbital y a la difenilhidantoína, fármacos algunas veces asociados en la terapia con otros anticonvulsivantes y con CBZ (Figura 3).
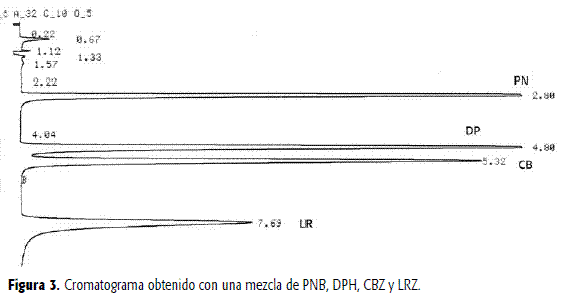
Para el caso de la Figura 3, las concentraciones empleadas de PNB, DPH, CBZ y LRZ fueron, respectivamente, 10, 10, 3 y 2 mcg/mL.
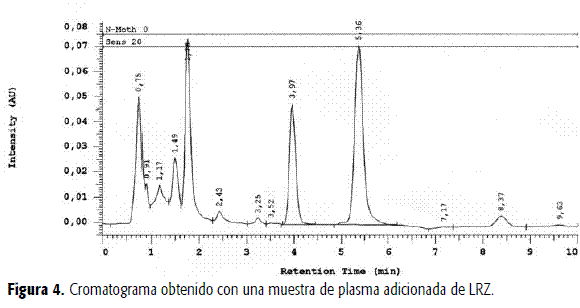
En la Figura 4 se presenta el cromatograma obtenido, en este caso empleando un equipo con detección mediante arreglo de diodos, con una muestra de plasma de un voluntario sano quien ingirió una dosis de CBZ, la cual se trató como se indica más adelante. Las señales entre 0 y 3,5 minutos corresponden a los componentes endógenos del plasma y metabolitos de CBZ. La pureza de las señales fue de 99,72% para CBZ y de 99,99% para LRZ, lo cual indica que la metodología es igualmente selectiva frente a los metabolitos de CBZ, siendo el principal el CBZ10-11 epóxido. En la figura los tiempos de retención fueron de 3,97 y 5,36 minutos para CBZ y LRZ, respectivamente. De acuerdo con los diferentes cromatogramas obtenidos con las muestras, los tiempos de retención relativos corresponden a 1 y 1,45 minutos para CBZ y LOR. Se observan, además, dos señales de baja intensidad, con tiempos de retención relativo de 1,3 y 0,56 minutos. La primera también se observa en el blanco del plasma, mientras que la segunda, que crece con el tiempo, podría corresponder al CBZ10-11 epóxido (13). El método se considera selectivo.
Linealidad
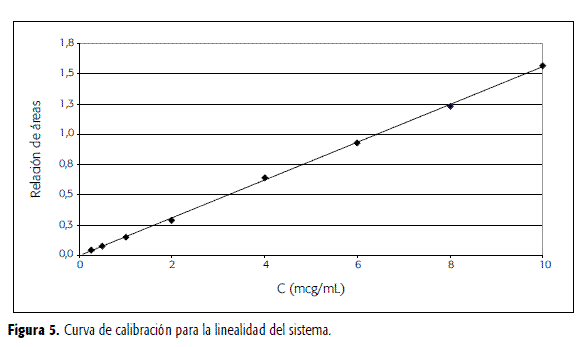
La curva de calibración para la linealidad del sistema se elaboró con concentraciones de CBZ entre 0,25 y 10 mcg/mL, cada una adicionada de LRZ en concentración 7,5 mcg/mL. La recta de regresión obtenida graficando la relación de áreas ÁreaCBZ/ÁreaLRZ contra la concentración de CBZ se presenta en la Figura 5.
Para la curva de linealidad del método, a volúmenes de 0,5 mL de plasma blanco se agregaron 75 mcL de una solución de LRZ 50 mcg/mL y volúmenes diferentes de una solución de CBZ 10 mcg/mL para obtener, luego de las etapas de lavado y extracción en fase sólida tal como se describió en el estudio de optimización, concentraciones finales de CBZ entre 0,25 y 10 mcg/mL. La concentración final de LRZ fue de 7,5 mcg/mL. Estudios previos demostraron que al disolver el residuo en metanol puro se obtenían los mismos resultados que al utilizar fase móvil. La gráfica para la linealidad del método se presenta en la Figura 6.
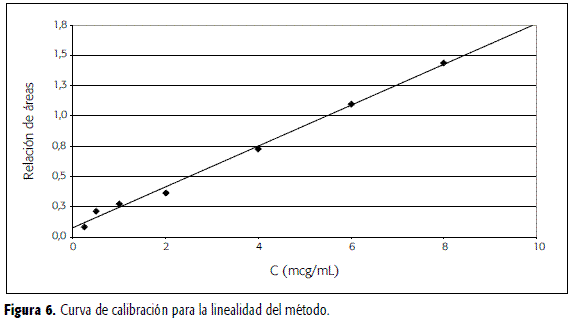
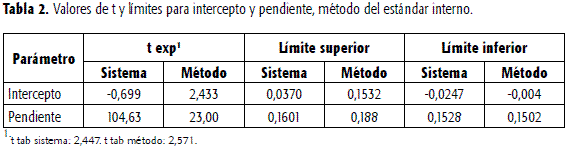
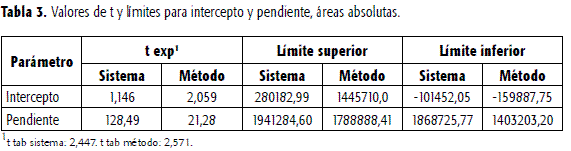
Las rectas se regresión y los test de Student para pendientes y para interceptos, indicaron para el sistema y el método pendientes significativamente diferentes de cero e interceptos no significativamente diferentes de cero. El origen de la curva se encuentra dentro de los límites de confianza para los interceptos. Mediante una prueba de F se encontró que las regresiones son significativas. En la Tabla 2 se presentan los valores de pendientes e interceptos, así como los correspondientes valores de t y F experimentales y los límites superior e inferior para ambos parámetros. El mayor valor de pendiente para la linealidad del método, se debe, como se mencionó, a que el LRZ se recupera en mayor proporción respecto a la CBZ. Igualmente, y por los resultados anteriores, interesaba trazar las rectas de regresión para el sistema y el método, teniendo en cuenta las áreas absolutas de CBZ. Los valores hallados para los parámetros de regresión se presentan en la Tabla 3. En este caso, la pendiente de la recta para el método es menor que la del sistema, e indicaría en principio una elusión de CBZ aproximada de 90%.
Las ecuaciones para las rectas de acuerdo con el ajuste por mínimos cuadrados obtenidas fueron:
Método estándar interno:
Sistema: Y = 0,15648 X - 0,0055, r2 = 0,9995
Método: Y = 0,1691 X + 0,0745, r2 = 0,9946
Áreas absolutas:
Sistema: Y = 1905005,19 X + 89365,47, r2 = 0,9998Método: Y = 1596045,8 X + 642911,1, r2 = 0,9927
Los valores de F experimentales en las dos rectas para cada una de las rectas son menores a los de F de la tabla para los correspondientes grados de libertad e indican que la regresión es significativa. Para el procedimiento del estándar interno, los valores experimentales fueron 10946,64 y 439,46 para el sistema y el método, respectivamente; valores superiores a 5,99 (gl = 1/6) y 6,61 (gl = 1/5), valores de la tabla para el sistema y el método, respectivamente.
Límites de detección y cuantificación
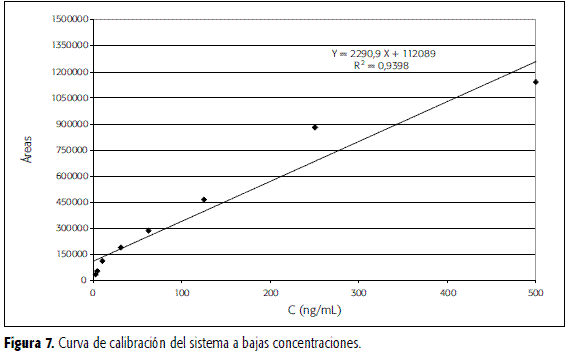
Teniendo en cuenta los bajos niveles de CBZ en plasma que se pueden en algún momento encontrar en plasma, se elaboró una nueva curva de calibración para ocho niveles de concentración de CBZ entre 2,5 y 500 ng/mL (sin adición de LRZ). La curva obtenida se presenta en la Figura 7. Las pruebas de t y de F indican regresión significativa e intercepto no significativamente diferente de cero. El límite de detección se puede establecer en 2,5 ng/mL y el de cuantificación en 8,25 ng/mL.
Precisión
La precisión se evaluó únicamente como repetibilidad para la CBZ a una concentración de 5 mcg/mL. Para seis determinaciones el coeficiente de variación obtenido fue de 1,48% y el área promedio 9508363,2 con límites superior e inferior de 9656152,6 y 9360573,3, respectivamente.
Exactitud
Para establecer el porcentaje de recuperación mediante el proceso de extracción en fase sólida de acuerdo con el procedimiento establecido, se estudiaron concentraciones de CBZ de 2, 4, 6 y 8 mcg/mL, adicionadas cada una de LRZ en concentración 7,5 mcg/mL. Para evaluar la recuperación, cada mezcla estándar se analizó antes y después de su paso por el cartucho de extracción.
La relación de áreas de las sustancias recuperadas, comparada con la relación obtenida a partir del análisis de las mezclas estándar, indica un porcentaje de recuperación promedio de 113.98% con una variación de 1,84%. Este valor se encuentra debido a la mayor recuperación de la CBZ respecto del LRZ. Al tener en cuenta los valores individuales de área antes y después del proceso de extracción se encuentran recuperaciones de 88,25% y 78,89% para CBZ y LRZ, respectivamente.
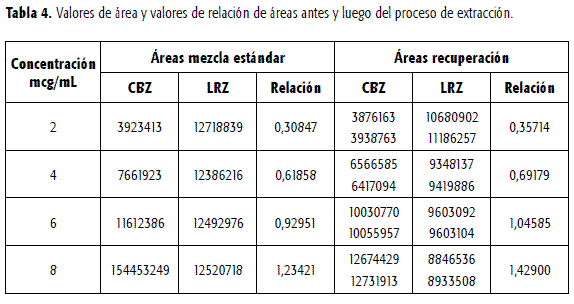
Un ensayo con plasma blanco adicionado de CBZ y LRZ para obtener concentraciones teóricas finales de 4 y 7,5 mcg/mL respectivamente, condujo a un porcentaje de recuperación de 112,3%.
Aplicación
La metodología validada para el análisis de CBZ en muestras de plasma se aplicó para su determinación en muestras provenientes de un voluntario sano, al cual se le administró en ayuno una dosis de CBZ, Tegretol® 200 mg. El voluntario fue privado de comida rica en grasas, de la ingesta de café, té, bebidas colas, bebidas alcohólicas, medicamentos y del uso de cigarrillo durante los tres días anteriores a la administración de la dosis, la cual se tomó con 250 mL de agua. Los resultados indican que la metodología puede ser aplicada con tal propósito. En las Figura 8, 9 y 10 se presentan los cromatogramas obtenidos luego del tratamiento y análisis según se especificó, de tres muestras de plasma correspondientes a tiempos de muestreo de 0,5, 6 y 24 horas. En la Tabla 5 se reportan los valores de área y su relación de CBZ y LRZ para las doce muestras de plasma tomadas a tiempos diferentes, así como la concentración de CBZ en cada muestra luego de interpolar en la curva de calibración del método.

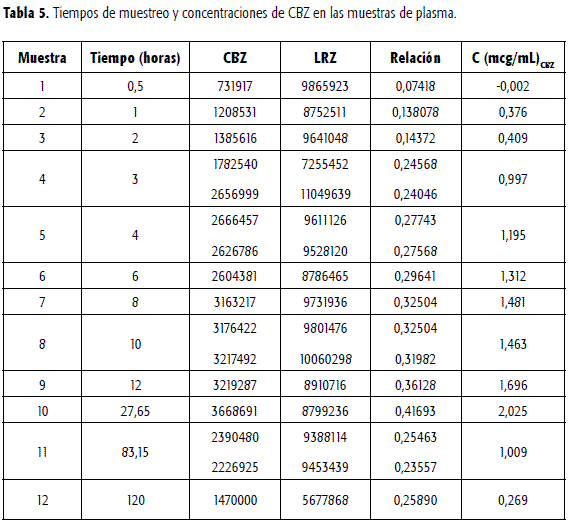
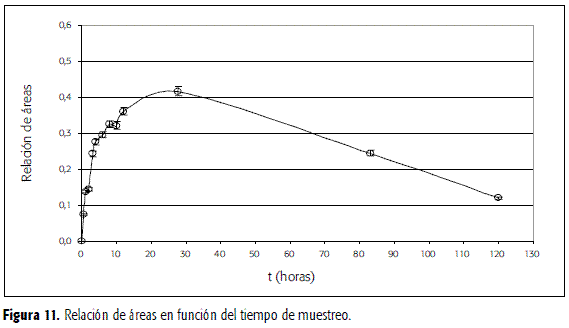
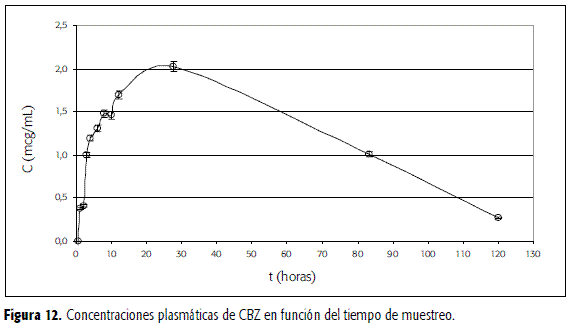
En las Figuras 11 y 12 se presentan, respectivamente, las curvas de relaciones de área y de concentración en plasma en función del tiempo de toma de la dosis de CBZ. Como se trabajó con un solo voluntario, de manera aproximada, se encuentra un tmax de 24 horas y un Cmax de 2,1 mcg/mL. Las concentraciones plasmáticas son con frecuencia máximas a las 8-10 horas de la toma, aunque ellas pueden retrasarse hasta 24 horas (2); dato éste último en concordancia con lo encontrado en este estudio. El reconocido efecto de primer paso que sufre la carbamazepina parece ser el responsable del comportamiento de la gráfica entre las 8 y las 10 horas.
CONCLUSIONES
Se desarrolló y se validó una metodología analítica para la determinación de carbamazepina en plasma por HLPC, método del estándar interno, en fase inversa utilizando una columna C18, una fase móvil compuesta por metanol-acetonitrilo-agua (20:25:55) a una temperatura de 30 °C y flujo de 1 mL/min. Como estándar interno se utilizó lorazepam. El proceso completo involucra un paso previo de extracción en fase sólida con cartuchos C18 antes del análisis cromatográfico. La metodología validada es selectiva, precisa y lineal entre un intervalo de concentraciones de 0,25 y 10 mcg/mL. La metodología es igualmente sencilla y conveniente para la determinación de niveles de carbamazepina en muestras de plasma de pacientes sometidos a tratamiento con este anticonvulsivante, niveles útiles para establecer la terapia más adecuada. Se aclara que en el caso de una terapia concomitante con difenilhidantoína, la resolución entre ésta y la carbamazepina no es completa. La metodología también puede ser utilizada en estudios de biodisponibilidad y equivalencia.
BIBLIOGRAFÍA
1. http://www.nlm.nih.gov/medlineplus/druginfo/uspdi/202111.html, julio 2006. [ Links ]
2. Goodman y Gilman, "Las bases farmacológicas de la terapéutica", editado por J.G. Hardman y L. Limbird, McGraw-Hill, décima edición, 2003, Vol. 1, pp. 541-542. [ Links ]
3. K.M. Patil y S.L. Bodhankar, Simultaneous determination of lamotrigine, phenobarbitone, carbamazepine and phenytoin in human serum by high-performance liquid chromatography, Journal of Pharmaceutical and Biomedical Analysis, 39, 181 (2005). [ Links ]
4. L.A. Saris, G. Brekelmans, G. van der Linden, R. Rademaker y P. Edelbroek, High-performance liquid chromatographic determination of carbamazepine and metabolites in human hair, Journal of Pharmaceutical and Biomedical Analysis, 41, 333 (2006). [ Links ]
5. R. Mandrioli, F. Albani, G. Casamenti, C. Sabbioni y M. Raggi, Simultaneous high-performance liquid chromatography determination of carbamazepine and five of its metabolites in plasma of epileptic patients, Journal of Chromatography B: Biomedical Sciences and Applications, 762, 109 (2001). [ Links ]
6. M. Gómez, M. Petrovic;, M. Fernández y D. Barceló, Determination of pharmaceuticals of various therapeutic classes by solid-phase extraction and liquid chromatography-tandem mass spectrometry analysis in hospital effluent wastewaters, Journal of Chromatography A., 1114, 224 (2006). [ Links ]
7. C. González, M. Lores, M. Casais y R. Cela, Simultaneous determination of neutral and acidic pharmaceuticals in wastewater by high-performance liquid chromatography-post-column photochemically induced fluorimetry, Journal of Chromatography A., 993, 29 (2003). [ Links ]
8. R. Chambers, Simultaneous determination by gas-liquid chromatography of carbamazepine and carbamazepine-10, 11-epoxide in plasma, Journal of Chromatography A., 154, 272 (1978). [ Links ]
9. D. Matthias y H. Wätzig, Purity control of carbamazepine by micellar electrokinetic chromatography, Journal of Chromatography A., 768, 113 (1997). [ Links ]
10. M. Raggi, V. Pucci, A. Maurizio, J. Muzikar y E. Kenndler, Separation of carbamazepine and five metabolites, and analysis in human plasma by micellar electrokinetic capillary chromatography, Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences, 770, 217 (2002). [ Links ]
11. Guidance for Industry, Bioanalytical Methods Validation, Draft Guiadance, Food and Drug Administration, Centre for Drug Evaluation and Research CDER, mayo 2001. [ Links ]
12. The United States Phamacopoeia, USP 29, The Pharmacopoeia Convention Inc., Rockville, USA, 2006, p. 588. [ Links ]
13. O.A. Quattrochi, R.F. Laba y S.I. Belaira, "Introducción a la cromatografía líquida de alta eficiencia, aplicación y práctica", Artes Gráficas Farro S.A., B.A., Argentina, 1992. [ Links ]
14. K. Chan, Simultaneous determination of carbamazepine and its epoxide metabolite in plasma and urine by high-performance liquid chromatography, Journal of Chromatography and Biomedical Application, 342, 341 (1985). [ Links ]

