










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
versión impresa ISSN 0034-7418
Rev. colomb. cienc. quim. farm. vol.42 no.2 Bogotá jul./dic. 2013
Artículo de investigación tecnológica
Estudio comparativo de la liberación in vitro de metformina, a partir de dos productos multifuente de liberación inmediata, comercializados en Colombia
Comparative study for in vitro release of metformin of two immediate-release multisource products, marketed in Colombia
Milena Pérez Guzmán1,2, Yenifer Orobio Lerma1, Yolima Baena Aristizábal2
1Centro Internacional de Entrenamiento e Investigaciones Médicas - cideim, carrera 125 No. 19-225, Cali, Colombia. Correo electrónico yorobio@cideim.org.co.
2CUniversidad Nacional de Colombia, Departamento de Farmacia, carrera 30 No. 45-03, Bogotá, D. C., Colombia. Correo electrónico: mrperezg@unal.edu.co, ybaenaa@unal.edu.co
Recibido para evaluación: 29 de julio de 2013.
Aceptado para publicación: 13 de septiembre de 2013.
RESUMEN
El presente estudio se realizó con el objetivo de establecer mediante pruebas de disolución in vitro, si dos formas farmacéuticas multifuente de liberación inmediata, de administración peroral con metformina como principio activo, son equivalentes en relación con su producto de referencia. Los datos de porcentaje de disolución obtenidos en tres niveles de pH (1,2; 4,5 y 6,8) y en dos equipos de disolución diferentes, se analizaron mediante el método de modelo independiente para comparar los perfiles de disolución de los productos a través del cálculo de los factores de diferencia (f1) y de similitud (f2), según las recomendaciones de la Food and Drug Administration (FDA). Para evaluar la cinética de liberación del fármaco, se aplicaron tres modelos matemáticos (ecuación de orden cero, de primer orden e Higuchi); además, para la comparación de los productos se aplicó un método basado en análisis multivariado para un diseño de medidas repetidas. Los resultados mostraron que existen diferencias estadísticamente significativas entre los productos, razón por la cual en este estudio no se demostró la equivalencia de los productos B y C con relación al producto A, con base en pruebas de disolución in vitro.
Palabras clave: metformina, multifuente, equivalencia, disolución in vitro, bioexención.
SUMMARY
The objective of the study was to determine the equivalence of two multisource drug products of metformin tablets using in vitro testing. The in vitro dissolution profiles were done in phosphate buffer (pH 6.8), acetate buffer (pH 4.5), hydrochloric acid (pH 1.2) and two dissolution apparatus were used. Multivariate analysis for repeated measures design was employed to compare the percentage dissolved of the drug products. According to Food and Drug Administration (FDA) guidance for dissolution data equivalence, model independent approach was applied. This involves the use of difference factor (f1) and similarity factor (f2). The drug release from drug products was analyzed by various mathematical models such as zero order, firs order and Higuchi. The results showed that there were significant differences between the drug products, and for this reason it was not possible to demonstrate equivalence based in vitro assays.
Key words:metformin, multisource, equivalence, in vitro dissolution, biowaiver
INTRODUCIÓN
Aunque los estudios de bioequivalencia han sido considerados como la prueba Gold Standard para establecer la intercambiabilidad de medicamentos, en 1995 Gordon Amidon y colaboradores, propusieron el Sistema de Clasificación Biofarmacéutica (bcs, por sus siglas en inglés) con el fin de facilitar el desarrollo de medicamentos de administración oral, reducir la necesidad de estudios de bioequivalencia y utilizar las pruebas de disolución in vitro como sustituto de estos estudios [1]. Este sistema clasifica los fármacos en cuatro categorías de acuerdo con su grado de solubilidad acuosa relativa a la dosis, y su permeabilidad a través de membranas biológicas, convirtiéndose en una de las herramientas predictivas más significativas de los últimos años, cambiando el paradigma de los estudios de bioequivalencia y siendo el tema de mayor debate regulatorio y científico de la última década [2]. Esta clasificación fue adoptada en Estados Unidos por la Food and Drug Administration (FDA), en Europa por la European Medicines Agency (ema) y en otros países por la World Health Organization (who) para ajustar los estándares de biodisponibilidad y bioequivalencia para medicamentos orales de liberación inmediata [2] y, posteriormente, es este marco científico la base de la guía Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System, emitida por la FDA en agosto de 2000.
Los medicamentos genéricos de liberación inmediata para uso sistémico deben demostrar bioequivalencia, pero hay excepciones para fármacos de clase I según el Sistema de Clasificación Biofarmacéutica (fármacos altamente solubles y altamente permeables) [3]; asimismo, si la disolución del medicamento genérico es similar a la del medicamento de referencia en tres medios de disolución de pH distinto (en un rango de 1 a 8), las pruebas de disolución in vitro se consideran suficientes para asegurar la bioequivalencia del producto [4]. Estos avances científicos han permitido a las agencias regulatorias de medicamentos de la mayoría de los países, la inclusión en su normatividad, de las propiedades biofarmacéuticas de los ingredientes farmacéuticamente activos y su aplicación como indicadores de biodisponibilidad, abreviando de esta manera los requerimientos para demostrar bioequivalencia y eximiendo de ensayos in vivo a gran cantidad de medicamentos. En este sentido, entre el 2000 y 2011, en Estados Unidos se hicieron 110 aprobaciones (42%) de fármacos de clase I con base en bioexenciones y en estudios de bioequivalencia [5]. Para este tipo de fármacos, cada vez hay más reportes de correlaciones entre las propiedades de solubilidad y permeabilidad con las pruebas de disolución cuando se aplican como sustitutos de ensayos de biodisponibilidad [6, 7, 8, 9, 10, 11, 12].
La FDA ha propendido por reducir el trámite regulatorio y los estudios innecesarios en humanos sin sacrificar la calidad de los productos [13]. Las pruebas de disolución han emergido como una muy importante herramienta en la industria farmacéutica de genéricos, especialmente para los procesos de formulación y desarrollo, en el monitoreo de procesos de manufactura y en pruebas de control de calidad; también son de gran utilidad para predecir el desempeño in vivo de ciertos productos. Las pruebas de disolución tienen un papel significativo al momento de tomar la decisión de requerir o no los estudios de bioequivalencia durante el escalamiento y cambios posaprobación (SUPAC) [13].
En los últimos años ha aumentado el interés de la comunidad científica, académica, de la industria farmacéutica y las entidades regulatorias en considerar a los productos con principios activos de la clase III (alta solubilidad y baja permeabilidad) según el Sistema de Clasificación Biofarmacéutica, como candidatos a la exención de estudios de bioequivalencia, teniendo como principal fundamento el hecho de que la etapa limitante en la velocidad de absorción para este tipo de fármacos, es la permeabilidad a través de la membrana intestinal y no la disolución [2, 14, 15]. Actualmente, las guías de la ema y de la who también plantean la exención de estudios de bioequivalencia para fármacos de clase III, si sus características de disolución in vitro son muy rápidas (= 85% de fármaco se disuelve en 15 minutos) [16]. Entre el 2000 y 2011, en Estados Unidos, el 37% de las aprobaciones realizadas con base en estudios de bioequivalencia, 98 correspondieron a fármacos de clase III, en su gran mayoría antidiabéticos [5].
Por las anteriores consideraciones, el fármaco evaluado en el presente estudio es la metformina, ya que por la naturaleza de sus propiedades biofarmacéuticas corresponde a un fármaco de clase III según el Sistema de Clasificación Biofarmacéutica (alta solubilidad y baja permeabilidad). Su cinética de absorción in vivo depende, principalmente, de la permeabilidad de la membrana y no tanto de la liberación del fármaco; por tanto, su disponibilidad es menos sensible a las características de disolución [4]. Algunos autores consideran que desde el punto de vista biofarmacéutico, la probabilidad de que los productos con metformina sean bioequivalentes es alta, si sus perfiles de disolución in vitro son similares y los excipientes no tienen efectos relevantes en la absorción, y con base en ello debería considerarse la bioexención para formas farmacéuticas perorales de liberación inmediata [17]. De acuerdo con esto, el presente estudio tiene como objetivo establecer si existe equivalencia in vitro entre dos formas farmacéuticas multifuente de liberación inmediata, de administración peroral con metformina como principio activo, y su correspondiente producto de referencia.
METODOLOGÍA
Materiales
Metformina HCl (estándar de referencia usp), metformina HCl (estándar secundario Sigma-Aldrich), fosfato monobásico de potasio (Scharlau), acetato de sodio (Sigma), ácido acético (Merck), ácido clorhídrico (Merck), hidróxido de sodio (Merck), agua purificada (obtenida a través de un sistema Milli-Q de Millipore).
Equipos
Espectrofotómetro UV/VIS (Genesys 6), equipo de disolución (TDT-08L Pharma Alliance Group), equipo de desintegración (Di-200 Pharma Alliance Group), equipo medidor de dureza (Dr. Schleuniger Pharmatron Model 5Y), balanza analítica (Ohaus serie Adventurer).
Pruebas de control de calidad
Antes de realizar las pruebas de disolución in vitro, los productos a evaluar se sometieron a la verificación de algunos aspectos técnicos como forma farmacéutica, contenido de principio activo, lote, fecha de fabricación, fecha de vencimiento, registro sanitario, condiciones e integridad del envase/empaque (envases o empaques primarios vacíos, rotos o mal sellados, con etiquetas húmedas, rasgadas o manchadas), concordancia entre la información del envase y del empaque, condiciones e integridad de la muestra, y características físicas y organolépticas (descripción, partículas extrañas, color, olor), además de los análisis fisicoquímicos correspondientes según la monografía de la usp 35, con el fin de verificar el cumplimiento de los requisitos de calidad en los productos: valoración de principio activo, uniformidad de unidades de dosificación, desintegración, dureza y disolución. Estas pruebas se realizaron por espectrofotometría UV a una longitud de onda de 233 nm.
Perfiles de disolución
Los productos evaluados en este estudio se seleccionaron teniendo en cuenta la presentación en tabletas de 850 mg de metformina clorhidrato. Se escogieron tres productos disponibles en el mercado: la formulación de referencia, a la cual se le asignó la letra A para efectos de identificación a lo largo de este estudio, y dos formulaciones de prueba de fabricación nacional, una que se comercializa en Colombia como producto de marca, a la que se le asignó la letra B, y una que se comercializa bajo la denominación genérica, a esta se le asignó la letra C. De cada uno de los productos a evaluar se adquirieron tres lotes de fabricación en establecimientos autorizados para la distribución y comercialización de medicamentos en la ciudad de Cali (Valle del Cauca). Las propiedades de disolución in vitro de los productos multifuente de metformina, frente al producto estándar de referencia correspondiente, se evaluaron bajo las condiciones recomendadas por la farmacopea vigente usp 35/nf30 y de acuerdo con los lineamientos de la Guía Dissolution testing of immediate release solid oral dosage forms [6], según lo muestra la (tabla 1).
Los perfiles de disolución se realizaron en doce unidades de cada producto a evaluar, con las condiciones de la usp 35, con tres medios de pH distintos y en dos equipos de disolución diferentes. El método analítico para la cuantificación de metformina en los tres medios de disolución se validó previamente, demostrando el cumplimiento de los criterios de linealidad, precisión, exactitud y especificidad, garantizando su aplicación en el presente estudio. En cada uno de los ocho vasos del equipo de disolución se adicionó de manera aleatoria, uno de los productos (producto A, B o C), en cinco ensayos independientes (en cinco días diferentes) para cada medio y para cada equipo de disolución. Los factores de tratamiento fueron los productos (A, B y C), el nivel de pH (6,8; 4,5 y 1,2) y el tipo de equipo (1 y 2). El factor de medidas repetidas fue el tiempo, la variable respuesta fue el promedio del porcentaje de disolución de las doce tabletas, y la unidad experimental fue la tableta. Se utilizó el promedio de las doce tabletas según las recomendaciones de la FDA, debido a que el coeficiente de variación fue menor del 20% para los primeros tiempos de muestreo (5, 10 y 15 minutos), y menor del 10% para los demás tiempos de muestreo (30, 45, 60, 90 y 120 minutos) [6].
De cada vaso de disolución se tomó una muestra a los 5, 10, 15, 30, 45, 60, 90 y 120 minutos, reponiendo el volumen tomado con medio fresco en cada muestreo; posteriormente se realizó una dilución de 1:100 con medio de disolución, y se obtuvieron lecturas por triplicado en el espectrofotómetro a una longitud de onda de 233 nm empleando como blanco, medio de disolución. Las tres lecturas se promediaron debido a que el coeficiente de variación entre ellas fue menor del 2%.
Evaluación del comportamiento de los perfiles de disolución
Entre los métodos más usados para comparar datos de disolución se encuentran los métodos estadísticos (método de análisis exploratorio de datos, diseño multivariado para medidas repetidas manova), el método del modelo dependiente (orden cero, primer orden, Higuchi, Hixson-Crowell, Weibull y modelo logístico) y el método de modelo independiente (factor de diferencia f1, factor de similitud f2, área bajo la curva, eficiencia de la disolución y tiempo medio de disolución) [18, 19]. El análisis realizado se fundamentó en las diferentes alternativas mencionadas y que son sugeridas por las guías de la FDA [6]. De esta manera, se empleó el método, modelo independiente, que se basa en el cálculo de los factores de diferencia (f1) y de similitud (f2) y se efectuó la descripción de los perfiles de disolución in vitro que incluye el cálculo de la eficiencia de la disolución y el tiempo medio de disolución [19, 45]. La eficiencia de la disolución (ed) se determinó a partir de los valores obtenidos en el área bajo la curva (abc) de los perfiles de disolución hasta 120 minutos, calculada por el método de los trapecios, y se expresó como porcentaje del área total del rectángulo definido por el 100% de disolución a 120 minutos [20, 21].
El método modelo dependiente se empleó para evaluar la cinética de liberación del fármaco mediante la aplicación de tres modelos matemáticos: ecuación de orden cero, de primer orden e Higuchi. Además, se realizó un análisis multivariado para un diseño de medidas repetidas con el fin de comparar los porcentajes de disolución de los productos bajo las condiciones establecidas.
RESULTADOS Y DISCUSIÓN
Pruebas de control de calidad
Empleando el método referenciado por la farmacopea de los Estados Unidos para metformina tabletas, se evaluaron las pruebas fisicoquímicas mencionadas anteriormente para los tres lotes de cada producto, a fin de verificar el cumplimiento de las especificaciones de calidad establecidas en la monografía oficial usp. Los resultados de los ensayos muestran que los tres lotes de cada producto (A, B y C) cumplieron las especificaciones de calidad de la usp 35 para valoración del principio activo (entre 95% y 105%) y uniformidad de unidades de dosificación (L1 menor a 15). Para la prueba de disolución, todos los productos cumplieron el criterio de la usp 35: mínimo 70% de fármaco debe disolverse en 45 minutos cuando se utiliza el aparato 1 (canastillas) a 100 rpm y buffer fosfato de potasio pH 6,8 como medio de disolución, y mínimo 75% de fármaco debe disolverse en 30 minutos cuando se utiliza el aparato 2 (paletas) a 75 rpm y buffer fosfato de potasio pH 6,8 como medio de disolución. El tiempo de desintegración fue de 12 minutos para los productos A y B, y de 10 minutos para el producto C.
Perfiles de disolución de metformina
En el marco de las consideraciones para la bioexención que deben cumplir los fármacos, se encuentra el criterio de rápida disolución, y para que un producto sea considerado de rápida disolución, mínimo el 85% de fármaco debe disolverse en 30 minutos usando los aparatos 1 o 2 recomendados por la farmacopea de los Estados Unidos y tres medios de disolución de pH distinto (ácido clorhídrico pH 1,2, buffer acetato de sodio pH 4,5 y buffer fosfato de potasio pH 6,8) [8]. La Figura 1, Figura 2 y Figura 3 permiten observar el comportamiento de los tres productos evaluados en los tres niveles de pH y en el equipo 1 (canastillas).
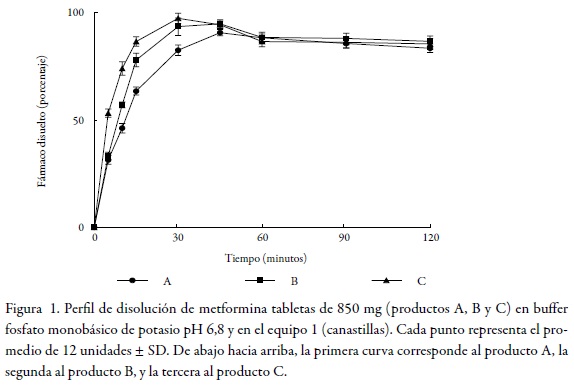
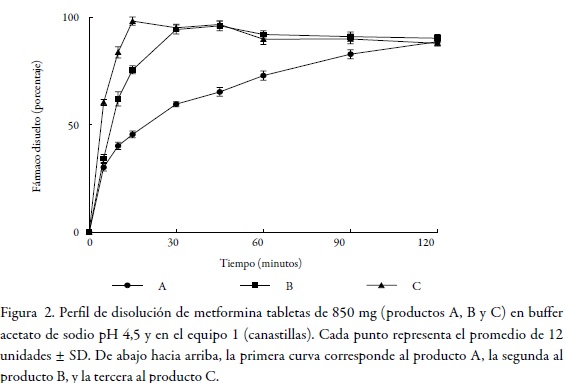
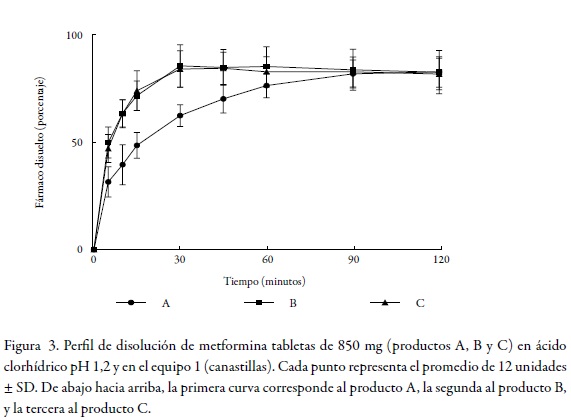
Dado que el criterio de rápida disolución de la FDA solo se cumplió para los productos B y C en pH 6,8 y en pH 4,5, pero no en pH 1,2 como se muestra en la tabla 2, tabla 3 dichos productos no pueden considerarse de disolución rápida. El producto A solo cumplió este criterio en buffer pH 6,8 y en el equipo 2; por tanto, tampoco puede ser considerado como un producto de rápida disolución.
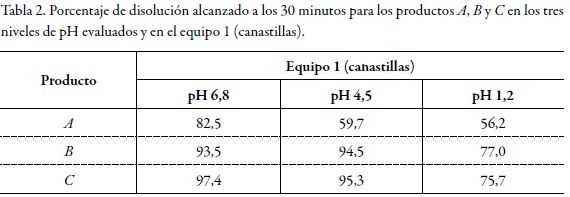
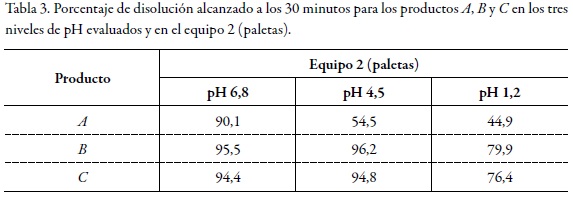
Los anteriores resultados evidencian que ninguno de los productos evaluados cumple este requisito para la solicitud de exención de estudios de bioequivalencia. No obstante, y con el propósito de aportar evidencia documentada al tema de bioexenciones para fármacos de clase III en el Sistema de Clasificación Biofarmacéutica, es necesario comparar el comportamiento de los perfiles de disolución de los productos B y C con relación al producto A, como se describe a continuación, a fin de determinar las relaciones de equivalencia entre ellos.
Evaluación del comportamiento de los perfiles de disolución
Según las recomendaciones de la FDA, si los productos a comparar muestran un porcentaje de disolución de al menos 85% en 15 minutos o menos, en los tres medios de disolución evaluados, el análisis mediante la aplicación del factor de similitud f2 no es necesario [8]. Como esto no se cumplió, la comparación de los perfiles de disolución por el método de modelo independiente es válida para este análisis, aunque se debe resaltar que el producto C en algunos casos presentó porcentajes de disolución mayores de 85% a los 15 minutos, pero no en los tres medios de disolución. Además, en este estudio, los perfiles de disolución de los productos se compararon aplicando el método de modelo dependiente y un método basado en análisis multivariado para un diseño de medidas repetidas, como se explicó en la metodología.
Análisis aplicando el método de modelo independiente
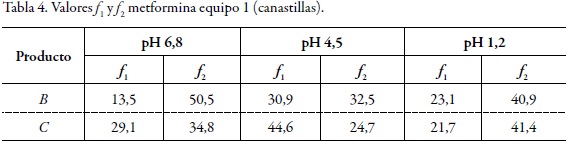
Dos perfiles de disolución se consideran similares si los valores de f1 están entre 0 y 15 y si los valores de f2 están entre 50 y 100 [6]. Teniendo en cuenta lo anterior y de acuerdo con los valores de f1 y f2 presentados en la tabla 4 para el equipo 1 (canastillas), solo el perfil de disolución del producto B presenta un comportamiento similar al producto de referencia (A) en buffer fosfato monobásico de potasio pH 6,8 como medio de disolución (f1 = 13,5 y f2 = 50,5), contrario a lo que ocurre en buffer acetato de sodio pH 4,5 y ácido clorhídrico pH 1,2 en los que los valores de f1 y f2 estuvieron fuera de los límites de aceptación. Se observa que los perfiles de disolución del producto C fueron diferentes al de referencia (A) en todos los medios de disolución. De acuerdo con los valores de f1 y f2 para la comparación de los productos B y C cuyas pruebas se realizaron en el equipo 2 (paletas), según la tabla 5 se observa que los perfiles de disolución de los productos B y C fueron diferentes al de referencia (A) en todos los medios de disolución (valores de f1 mayores a 15 y valores de f2 menores de 50).

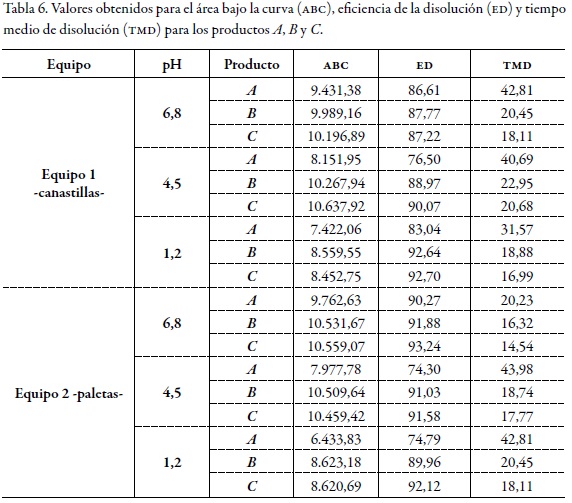
La aplicación del método de modelo independiente incluye el cálculo de la eficiencia de la disolución (ed) y del tiempo medio de disolución (tmd). La ed permite la comparación de varias formulaciones simultáneamente y puede ser teóricamente relacionada con la curva de concentración plasmática vs. tiempo obtenido mediante técnicas de deconvolución de datos in vivo [22]. El tiempo medio de disolución se calculó con el fin de caracterizar la velocidad de disolución del fármaco. En formas farmacéuticas de liberación inmediata, este parámetro indica el tiempo promedio requerido para la disolución del fármaco [23], y es de gran utilidad en formas farmacéuticas de liberación sostenida para determinar el efecto retardante que ejerce, por ejemplo, un polímero [24]. Los resultados de la tabla 6 evidencian que el producto de referencia (A) es el que presenta el mayor valor para estos parámetros en comparación con los productos B y C; especialmente, los valores del tiempo medio de disolución (tmd) de los productos B y C son considerablemente menores al producto A.
Análisis aplicando el método de modelo dependiente
Para evaluar la cinética de liberación de metformina en los productos en estudio, se aplicaron tres modelos matemáticos: ecuación de orden cero, de primer orden e Higuchi. De acuerdo con los valores de r2 presentados en la tabla 7, se observa que la liberación del fármaco en los productos evaluados se ajusta a una cinética de primer orden en pH 6,8 y en el equipo 1. Este modelo sugiere que la velocidad de liberación depende de la concentración de fármaco disuelto. Las formas farmacéuticas que siguen este perfil de disolución, tales como las que contienen los fármacos solubles en agua en matrices porosas, liberan el fármaco de manera proporcional a la cantidad de principio activo remanente en su interior, de tal manera que la cantidad liberada por unidad de tiempo disminuye [25]. A pH 4,5 y 1,2, en el mismo equipo, el comportamiento de los productos se ajustó mejor al modelo de difusión de Higuchi (a excepción del producto B cuyo mejor ajuste fue al de una cinética de primer orden en pH 4,5). En el equipo 2, los productos se ajustaron mejor al modelo de Higuchi a pH 6,8 y 4,5 (a excepción de los productos A y C cuyo mejor ajuste fue al de una cinética de primer orden en pH 6,8 y 4,5, respectivamente). A pH 1,2 los productos se ajustaron mejor al modelo de primer orden (a excepción del producto A cuyo mejor ajuste fue al del modelo de Higuchi). Higuchi describe la liberación de fármacos como un proceso de difusión basado en la Ley de Fick, dependiente de la raíz cuadrada del tiempo. Esta relación puede ser usada para describir la disolución del fármaco a partir de formas farmacéuticas de liberación modificada, como en el caso de los sistemas de liberación transdérmica y matrices con fármacos solubles en agua [25].

Con base en los valores de la constante de velocidad de disolución del modelo de primer orden (K1) e Higuchi (KH) presentados en la tabla 7, se evidencia que el producto A presenta la menor velocidad de disolución en los tres medios evaluados. Lo anterior refleja la influencia que pueden tener las condiciones de disolución (medio de disolución y pH, tipo de equipo, velocidad de agitación), no solo sobre la velocidad de disolución, sino también sobre la cinética de liberación de los fármacos [26, 27].
Existen varios estudios realizados con el objetivo de caracterizar la liberación del fármaco a partir de diferentes formulaciones de metformina, en las que se varían las proporciones de los auxiliares de formulación con el fin de obtener una liberación controlada, sostenida o extendida. Los resultados de dichos estudios muestran que la velocidad de liberación del fármaco puede verse afectada por los componentes de la formulación, así como por el método de fabricación del producto [10, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45]). Los excipientes pueden afectar la velocidad y la extensión de la absorción, y es por ello por lo que la FDA incluye este tema como uno de los aspectos para tener en cuenta al momento de solicitar la exención de estudios de bioequivalencia, a la vez que recomienda que cuando en la formulación se empleen nuevos excipientes o excipientes ya aprobados por la FDA pero en cantidades considerablemente diferentes a las comúnmente usadas, se debe documentar mediante estudios in vivo la ausencia de impacto en la biodisponibilidad del fármaco [8]. Debido a que se desconoce la composición de los productos evaluados, no fue posible determinar el efecto de los auxiliares de formulación sobre las características de disolución de los productos.
Análisis aplicando el método basado en análisis multivariado para un diseño de medidas repetidas
En este estudio se realizó el análisis para un diseño de medidas repetidas, dado que las unidades experimentales manifiestan cambios a través del tiempo en respuesta a varios factores controlados por el investigador. Este diseño considera el tiempo como el factor de repetición y el porcentaje disuelto como la variable dependiente [18]. Los análisis se realizaron en el programa estadístico IBM SPSS 20.0, con el fin de contrastar el porcentaje de disolución de los productos en los diferentes tiempos de medición bajo los tratamientos aplicados (equipo y nivel de pH).
- Tipo de equipo: existe evidencia estadística para no rechazar • la hipótesis que plantea que los porcentajes de disolución promedio obtenidos en los equipos canastillas y paletas son iguales. Esto indica que el porcentaje de disolución promedio no presenta diferencias al cambiar de equipo de observación. Los intervalos de confianza del 95% llevan a la misma conclusión.
- Nivel de pH: se rechazó la hipótesis que plantea que los porcentajes de disolución promedio obtenidos a los niveles de pH observados son iguales, lo que significa que se presentan diferencias significativas al variar el nivel de pH. La comparación múltiple entre los pares del nivel de pH ajustado se hizo mediante la corrección de Bonferroni; además, los intervalos de confianza reforzaron esta conclusión.
- Producto: para la comparación de los productos, se tomó el producto de referencia A como grupo de comparación y se efectuó el ajuste de Dunnet (t de dos colas), y se encontró, por tanto, que a un nivel de significancia de 0,05 existen diferencias estadísticamente significativas entre el producto de referencia A y el producto genérico B, así como entre el producto A y el producto genérico C.
Aunque los resultados del presente estudio muestran que los productos B y C no son similares al producto de referencia (A) a través de la comparación in vitro de los perfiles de disolución aplicando los factores f1 y f2 recomendados por la FDA, dichos resultados aportan información a la discusión ya existente sobre bioexención para fármacos con clasificación biofarmacéutica tipo III y merecen ser analizados con mayor detalle en fases posteriores a fin de determinar los efectos relevantes que puedan estar ejerciendo los auxiliares de formulación presentes en los productos evaluados sobre la disolución y, por tanto, sobre la absorción, en un rango de pH fisiológico.
CONCLUSIONES
Teniendo en cuenta los resultados obtenidos en el presente estudio, se observan considerables diferencias entre los perfiles de disolución de los productos B y C comparados con el producto de referencia (A); se resalta este último como el producto con el menor desempeño en cuanto a la velocidad de liberación del fármaco (ni a los 15 ni a los 30 minutos alcanza un porcentaje de liberación de 85%), área bajo la curva, eficiencia de la disolución y tiempo medio de disolución. Debido a estas características de desempeño, las cuales evidentemente son diferentes en los productos evaluados, los productos mul tifuente B y C no pueden ser considerados equivalentes con base en los resultados de este estudio in vitro, a pesar de tener un desempeño superior en los parámetros evaluados..
AGRADECIMIENTOS
Al Centro Internacional de Entrenamiento e Investigaciones Médicas (cideim), por el apoyo financiero para la realización de la "Maestría en Ciencias Farmacéuticas" en cuyo plan de estudios se desarrolló la presente investigación. De manera especial, a la doctora Nancy Gore Saravia, directora científica de CIDEIM.
REFERENCIAS
1. M. Yasir, M. Asif, A. Kumar, A. Aggarval, Biopharmaceutical 1. classification system: An account, International Journal of PharmTech Research, 2(3), 1681 ( [ Links ]2010).
2. A. Dahan, J.M. Miller, G. Amidon, Prediction of solubility and permeability class membership: Provisional bcs classification of the world´s top oral drugs, The aaps Journal, 11(4), 740 (2009) [ Links ].
3. G.L. Amidon, H. Lennernas, V. Shah, J.A. Crison, Theorical basis for a Biopharmaceutical Drug Classification: The correlation of in vitro drug product dissolution and in vivo bioavailability, Pharmaceutical Research, 12(3), 413 (1995) [ Links ].
4. I. Homsek, J. Parojcic, M. Dacevic, L. Petrovic, D. Jovanovic, Justification of metformin hydrochloride biowaiver criteria based on bioequivalence study, Arzneimittelforschung, 60(9), 553 (2010) [ Links ].
5. A.K. Nair, O. Anand, N. Chun, D.P. Conner, M.U. Mehta, D.T. Nhu, et al., Statistics on bcs classification of generic drug products approved between 2000 and 2011 in the usa, The aaps Journal, 14(4), 664 (2012) [ Links ].
6. FDA Guidance for Industry. Dissolution testing of immediate release solid oral dosage forms. usa Department of Health and Human Services. Food and Drug Administration Center for Drug Evaluation and Research (cder), 1997. Disponible en http://www.FDA.gov/downloads/Drugs/GuidanceComplianceRegulatory- Information/Guidances/ucm070237.pdf. Acceso el 17 de mayo de 2013. [ Links ]
7. who Multisource (generic) pharmaceutical products: Guidelines on registration requirements to establish interchangeability, 2005. Disponible en http://www. who.int/medicines/services/expertcommittees/pharmprep/QAS04_093Rev4_final. pdf. Acceso el 19 de febrero de 2013. [ Links ]
8. FDA Guidance for Industry. Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a Biopharmaceutic Classification System. usa, Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (cder). Disponible en www.FDA.gov/cder/guidance/3618fnl.htm. Acceso el 8 de mayo de 2013. [ Links ]
9. FDA Guidance for Industry. Bioavailability and bioequivalence studies for orally administered drug products-general considerations. usa, Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (cder). Disponible en http://www.FDA.gov/cder/guidance/ index.htm. Acceso el 28 de abril de 2013. [ Links ]
10. who. Schedule for the adoption process of document: Proposal to waive in vivo bioequivalence requirements for the who model list for essential medicines immediate release, solid oral dosage forms, oct. 2005. Disponible en http://www. who.int/medicines/services/expertcommittees/pharmprep/QAS04_109Rev1_ Waive_invivo_bioequiv.pdf. Acceso el 12 de diciembre de 2012. [ Links ]
11. The European Agency for the Evaluations of Medicinal Products. Evaluation of medicines for human use. Disponible en http://eudra.org/emea.html. Acceso el 21 de marzo de 2013 [ Links ]
12. M. Martínez, L. Augsburger, T. Jhonston, W. Warren, Applying the Biopharmaceutics Classification System to Veterinary Pharmaceutical Products. Part I: Biopharmaceutics and formulation considerations, Advanced Drug Delivery Reviews, 54, 805 (2002) [ Links ].
13. O.M. Anand, L.X. Yu, D.P. Conner, B.M. Davit, Dissolution testing for generic drugs: An FDA perspective, The aaps Journal, 13(3), 328 (2011) [ Links ].
14. J.E. Polli, B. Abrahamsson, L.X. Yu, G.L. Amidon, J.M. Baldoni, J.A. Cook, et al., Summary workshop report: Bioequivalence, Biopharmaceutics Classification System, and beyond, The aaps Journal, 10(2), 373 (2008) [ Links ].
15. S. Stavchansky, Scientific perspective on extending the provision for waivers of in vivo bioavailability and bioequivalence studies for drug products containing high solubility-low permeability drugs (bcs-Class 3), The aaps Journal, 10(2), 300 (2008) [ Links ].
16. L. Chen, V.P. Shah, D.J. Crommelin, L. Shargel, D. Bashaw, M. Bhatti, et al., Harmonization of regulatory approaches for evaluating therapeutic equivalence and interchangeability of multisource drug products: Workshop summary report, The aaps Journal, 13(4), 556 (2011) [ Links ].
17. C.L. Cheng, L.X. Yu, H.L.Lee, C.Y. Yang, C.S. Lue, C.H. Chou, Biowaiver extension potential to bcs class III high solubility-low permeability drugs: Bridging evidence for metformin immediate-release tablet, European Journal of Pharmaceutical Sciences, 22(4), 297 (2004) [ Links ].
18. E. Demirtürk, O. Levent, Evaluation of in vitro dissolution profile comparison methods of immediate release gliclazide tablet formulations, Hacettepe University, Journal of Faculty of Pharmacy, 25(1), 1 (2005) [ Links ].
19. L. Bajerski, R. Cassanta, C. Dias, A.M. Bergold, P.E. Fröehlich, Development and validation of a discriminating in vitro dissolution method for a poorly soluble drug, olmesartan medoxomil: Comparison between commercial tablets, aaps PharmSciTech, 11(2), 637 (2010) [ Links ].
20. S. Poongothai, V. Balaji, B. Madhavi, A.R. Rajasekhar, R. Llavarasan, C.M. Karrunakaran, A sensitive dissolution test method for the development and validation of levetiracetam tablets by reverse phase-HPLC technique, International Journal of PharmTech Research, 3(2), 1023 (2011) [ Links ].
21. B.A. Alkhalidi, H.S. Alkhatib, A.A. Khdair, Comparative dissolution of diltiazem immediate and extended release products using conventional usp and innovative dissolution paddles, The Open Drug Delivery Journal, 4, 48 (2010) [ Links ].
22. S. Yamamura, F. Aida, Y. Momose, E. Fukuoka, Analysis of mean disintegration time and mean dissolution time by moment analysis using microcalorimetric curves, Drug Development and Industrial Pharmacy, 26(1), 1 (2000) [ Links ].
23. K.J. Wadher, R.B. Kakde, M.J. Umekar, Development of a sustained-release tablet of metformin hydrochloride containing hydrophilic eudragit and ethyl cellulose polymer, International Journal of Comprehensive Pharmacy, 2(5), 1 (2011) [ Links ].
24. P. Costa, J.M. Sousa, Modeling and comparison of dissolution profiles, European Journal of Pharmaceutical Sciences, 13, 123 (2001) [ Links ].
25. Disolution testing for polyoxTM extended release matrices. Disponible en http://www.colorcon.com. Acceso el 29 de abril de 2013. [ Links ]
26. L.F. Ponde D´León, L.A. Oviedo, J.M. Aiache, Análisis del efecto del pH en la cinética de liberación de teofilina en un medicamento de liberación programada: Parte I. Microgránulos encapsulados recubiertos con goma laca, Revista Colombiana de Ciencias Químico-Farmacéuticas, 34(2), 126 (2005) [ Links ].
27. K.J. Wadher, R.B. Kakde, M.J. Umekar, Effect of hydroxypropyl methylcellulose and ethyl cellulose polymer on release profile and kinetics of metformin HCl from matrix tablets, International Journal of PharmTech Research, 3(3), 1850 ( [ Links ]2011).
28. R. Narasimharao, M. Anusha Reddy, N. Swetha Reddy, P. Divyasagar, K. Keerthana, Design and evaluation of metformin hydrochloride extended release tablets by direct compression, International Journal of Research in Pharmaceutical and Biomedical Sciences, 2(3), 1118 (2011) [ Links ].
29. R. Margret Chandira, A.A. Mohamed Yasir Arafath, D. Bhowmik, B. Jayakar, K.P. Sampath Kumar, Formulation and evaluation of bilayered floating tablets of metformin hydrochloride, The Pharma Innovation, 1(6), 26 (2012) [ Links ].
30. R. Raparla, E.G. Krishna Murthy Talasila, Design and evaluation of floating drug delivery systems of metformin with natural gums as release retarding polymers, International Journal of Advances in Pharmaceutics, 1(1), 22 (2012) [ Links ].
31. J. Rojas, C. González, C. Rico, O. Sáez, Formulation of a modified release metformin HCl matrix tablet: Influence of some hydrophilic polymers on release rate and in vitro evaluation, Brazilian Journal of Pharmaceutical Sciences, 47(3), 483 ( [ Links ]2011).
32. S. Basu Ghosh, S. Mukherjee, Design, fabrication and in vitro evaluation of metformin HCl matrix tablets for treatment of diabetes mellitus, Science and Technology, 2(6), 168 (2012) [ Links ].
33. P. Divakar, D. Kumar, C. Praveen, C. Sowmya, C. Suryaprakash Reddy, Formulation and in vitro evaluation of liposomes containing metformin hydrochloride, International Journal of Research in Pharmaceutical and Biomedical Sciences, 4(2), 479 (2013) [ Links ].
34. tablets of metformin hydrochloride, International Journal of Pharmaceutical, Chemical and Biological Sciences, 2(3), 401 (2012) [ Links ].
35. K.J. Wadher, R.B. Kakde, M.J. Umekar, Formulation of sustained release metformin hydrochloride matrix tablets: Influence of hydrophilic polymers on the release rate and in vitro evaluation, International Journal of Research in Controlled Release, 1(1), 9 (2011) [ Links ].
36. K. Kumari, C. Sharma, P.P. Kundu, In vitro release of metformin hydrochloride from films of chitosan-methylcellulose blends, Asian Journal of Chemistry, 21(10), 148 (2009) [ Links ].
37. A.M. Olusola, A.L. Adekoya, O.J. Olanrewaju, Comparative evaluation of physicochemical properties of some commercially available brands of metformin HCL tablets in Lagos, Nigeria, Journal of Applied Pharmaceutical Science, 2(2), 41 (2012) [ Links ].
38. L.C. Block, L.O. Schemling, A.G. Couto, S.C. Mourão, T.M.B. Bresolin, Pharmaceutical equivalence of metformin tablets with various binders, Rev. Ciênc. Farm. Básica Apl., 29(1), 29 (2008) [ Links ].
39. R. Diwedi, A. Alexandar, M.J.N. Chandrasekar. Preparation and in vitro evaluation of sustained release tablet formulations of metformin HCl, Asian Journal of Pharmaceutical and Clinical Research, 5(1), 45 (2012) [ Links ].
40. S.A. Afifi, S. Ahmadeen, A comparative study for evaluation of different brands of metformin hydrochloride 500 mg tablets marketed in Saudi Arabia, Life Science Journal, 9(4), 4260 (2012) [ Links ].
41. A. Roy, K. Roy, S. Roy, J. Deb, A. Ghosh, K. Asraf Ali, Response surface optimization of sustained release metformin-hydrochloridematrix tablets: Influence of some hydrophillic polymers on the release, International Scholarly Research Network Pharmaceutics, 1 (2012) [ Links ].
42. M. Charde, S. Jayani, D. Pandey, R.D. Chakole, Formulation and evaluation of immediate release tablets of metformin hydrochloride on laboratory scale, International Journal of Advances in Pharmaceutical Analysis, 1(2), 45 (2011) [ Links ].
43. H. Roy, C.K. Brahma, S. Nandi, K.R. Parida, Formulation and design of sustained release matrix tablets of metformin hydrochloride: Influence of hypromellose and polyacrylate polymers, International Journal of Applied and Basic Medical Research, 3(1), 55 (2013) [ Links ].
44. I. Kocic, I. Homsek, M. Dacevic, J. Parojcic, B. Miljkovic, 44. An investigation into the influence of experimental conditions on in vitro drug release from immediate- release tablets of levothyroxine sodium and its relation to oral bioavailability, aaps PharmSciTech, 12(3), 938 (2011) [ Links ].
45. N. Yuksel, A.E. Kanik, T. Baykara, Comparison of in vitro dissolution profiles by anova-based, model-dependent and -independents methods, International Journal of Pharmaceutics, 209, 57 (2000) [ Links ].
