




![Evaluación de la actividad anticonvulsiva de pirazolo [1,5-a] [1,3,5] triazinas obtenidos por síntesis](/img/es/next.gif)





Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
versión impresa ISSN 0034-7418
Rev. colomb. cienc. quim. farm. vol.43 no.1 Bogotá ene./jun. 2014
https://doi.org/10.15446/rcciquifa.v43n1.45461
http://dx.doi.org/10.15446/rcciquifa.v43n1.45461
Artículo de investigación clíca
Desarrollo y validación de una metodología por cromatografía líquida de alta eficiencia para la determinación simultánea de piperacilina sódica y tazobactam sódico en inyectables para uso humano
Validation of a high performance liquid chromatography method for the simultaneous determination of sodium piperacillin and sodium tazobactam in human injections
Noralba Sierra1, Gustavo Gómez2
1Departamento de Farmacia, Universidad Nacional de Colombia, A.A.14490, Bogotá D. C., Colombia. Correo electrónico: nsierram@unal.edu.co
2Laboratorios Sumimed, Bogotá D. C., Colombia.
Recibido para evaluación: Septiembre 2 de 2013.
Aceptado para publicación: Mayo 15 de 2014.
RESUMEN
Se presentan en este artículo los resultados obtenidos en desarrollo y validación de una metodología analítica para la cuantificación simultánea de piperacilina sódica y tazobactam sódico en inyectables para uso humano. El procedimiento consiste en una separación por cromatografía líquida de alta eficiencia (HPLC) en fase inversa empleando como fase móvil una mezcla de acetonitrilo e hidróxido de tetrabutilamonio 0,005M de pH 5,0 (31:69), una columna C18 a 38°C y detección a una longitud de onda de 220 nm. Se encontró que el método es selectivo, lineal y preciso. Estas características junto con su sencillez permiten que el método sea adecuado y conveniente para el objetivo propuesto. La robustez de la metodología fue también investigada. El método validado se aplicó para la determinación de las dos sustancias en un producto inyectable del mercado colombiano con registro sanitario del Instituto Nacional de Vigilancia y Control de Medicamentos y Alimentos (Invima).
Palabras clave:Cromatografía líquida - Detección ultravioleta - Piperacilina sódica - Tazobactam sódico - Validación.
SUMMARY
A reverse phase high performance liquid chromatographic method was developed for the simultaneous quantitative assay of sodium piperaciline and sodium tazobactam. The method was validated and then applied to the quantitative determination assay of each of two substances A C18 column stabilized at 38 °C was used and the detection was performed at 220 nm. A mixture of acetonitrile and tetrabutylammonium hydroxide 0,005M pH 5,0 (31:69) was used as mobile phase. The method is selective, linear and shows a good repeatibility. The robustness was also verified. These properties besides their simplicities make it convenient for the objective proposed. The validated method was applied for the simultaneous assay of piperaciline and tazobactam in one market products with sanitary register at Instituto Nacional de Vigilancia y Control de Medicamentos y Alimentos (INVIMA).
Key words:Reverse high performance liquid chromatography - Sodium piperaciline - Sodium tazobactam - UV detection - Validation.
INTRODUCCIÓN
La piperacilina es un antibiótico betaláctamico de amplio espectro, efectivo contra una gran variedad de microorganismos, incluyendo aquellos resistentes a la penicilina como el Streptococcus pneumoniae [1]. Es común encontrar este antibiótico asociado con inhibidores de la beta-lactamasa, tal como tazobactam, con lo cual se pretende aumentar la actividad de la piperacilina frente a microorganismos productores de la enzima. Este efecto sinérgico fue demostrado por Zaghloul y colaboradores [2], empleando una combinación de ocho partes de piperacilina por una parte de tazobactam. Diferentes estudios se han realizado para comparar la actividad de algunas cefalosporinas y antibióticos betalactámicos, con la presentada por la asociación de piperacilina y tazobactan [1, 3-5]. Diferentes métodos de análisis se encuentran en la literatura para la determinación de piperacilina y de tazobactam en muestras de variada naturaleza, así como para su determinación en mezclas. Peyrin y colaboradores determinan tazobactam, previa derivatización con 1,2,4-triazol [6] en suero y fluído hemofiltrado, alcanzando límites de detección del orden de 50 ng/mL. Por otra parte, Tang y colaboradores [7] realizan un estudio sobre las propiedades fluorescentes y su aplicación de los productos de descomposición de la piperacilina. Pajchel y Tyski [8] determinan piperacilina en presencia de tazobactam mediante electroforesis capilar micelar, empleando dodecilsulfato de sodio como agente formador de micelas, comparando los resultados obtenidos por cromatografía líquida. Marcelos y Archontakis [9] determinan por cromatografía líquida piperacilina y tazobactam en un polvo inyectable, utilizando una columna C18 y una fase móvil de pH 5,0, compuesta por fosfato monosódico, acetonitrilo y metanol. La literatura reporta igualmente la determinación de piperacilina y tazobactam en muestras de orina y plasma utilizando una columna C18 y una fase móvil compuesta por acetonitrilo y acetato de amonio [10]. Utilizando elusión con gradiente y supresión iónica, Ocampo y colaboradores determinan, sobre una columna C18 y detección ultravioleta, las dos sustancias en muestras de plasma, suero, bilis y orina [11]. La determinación de piperacilina y tazobactam mediante HPLC de par iónico en materiales biológicos ha sido igualmente estudiada [12].
En el presente artículo se muestran los resultados obtenidos en el desarrollo y validación de un método de análisis cuantitativo para piperacillina sódica y tazobactan sódico por HPLC en fase inversa, empleando una columna de 125 x 4,5 mm con octadecilsilano como fase estacionaria en la modalidad de par iónico y detección por espectrofotometría ultravioleta a 220 nm. Los niveles de concentración de piperacilina y de tazobactam utilizados en la validación de los diferentes parámetros, se establecieron teniendo en cuenta la composición del producto sobre el cual se aplicará la metodología validada, 8 partes de piperacilina por 1 parte de tazobactam, tanto materia prima estéril como inyectable.
Para el desarrollo de la metodología analítica se partió de la documentación entregada por la compañía FADA (Argentina) a través de Sumimed (Colombia) para la determinación en forma separada de las dos sustancias, con dos sistemas cromatográficos diferentes. Igualmente se consultó la monografía de la Farmacopea Europea [13] para el control de calidad de piperacilina sódica, materia prima e inyectables. La farmacopea americana USP 35-NF 30 [14] reporta la determinación de los dos principios activos por HPLC con una columna fenilo de uso poco extendido. Como fase móvil emplea una mezcla de acetonitrilo - hidróxido de tetrabutilamonio 0,4M de pH 3,8 (25:75). Como propósito fundamental se fijó el desarrollar una metodología única para la determinación simultánea de las dos sustancias con una columna C18, disponible en la mayoría de los laboratorios de análisis de medicamentos, y una fase móvil compuesta por una mezcla de acetonitrilo e hidróxido de tetrabutilamonio a pH 5,0 con menor concentración de éste y por consiguiente menor deterioro de la fase estacionaria. Esta menor concentración es suficiente para obtener señales simétricas. En principio podría observarse una dificultad por la relación de concentraciones en el producto, la cual podría obviarse si se demuestra linealidad dentro de un intervalo adecuado de concentraciones para cada uno de los dos fármacos, manteniendo siempre la mencionada relación.
METODOLOGÍA
Equipos y materiales
Cromatográfo líquido Waters 510, detector UV Waters Ass. 484, integrador Perkin Elmer LCI 100, inyector Rheodyne 20 microlitros., columna LiChrospher Merck RP 18 (C18), 4,5x 125mm, 5 µm, bomba cuaternaria Perking Elmer Serie 410, cámara ambiental de solventes Sec-4 Perking Elmer, horno cromatográfico LC-100 Perking Elmer, integrador LCI 100 Perking Elme, detector UV-VIS Water, baño ultrasónico Erma, espectrofotómetro UV/VIS Unicam UV2.
Sustancias y reactivos
Tazobactam estándar secundario 99,3%, piperacilina sódica estándar secundario 95,9% (90,2% como base), materia prima piperacilina + tazobactam (8:1), acetonitrilo HPLC Merck, hidróxido de tetrabutilamonio (HTBA) 20% RA Merck, ácido fosfórico al 85% p/p RA Merck, filtros para muestras 0,45 µm, membranas para filtración de solventes 0,45 µm, sistema de filtración mediante vacío Millipore para líquidos.
Estandarización de la metodología
Para el establecimiento de los parámetros de validación se siguieron las pautas recomendadas por la Farmacopea Americana USP 35, Categoría I [15] y otras reportadas en la literatura [16-20]. Como se mencionó, para cada parámetro de validación, las concentraciones empleadas de tazobactam y de piperacilina guardan relación con las concentraciones etiquetadas en el producto mezcla de los mismos.
Optimización del sistema cromatográfico
Para la estandarización de la metodología analítica se realizaron varios y diferentes ensayos en procura de obtener las mejores condiciones cromatográficas para la determinación de las dos sustancias en estudio. En esta etapa, se realizaron varios ensayos para ajustar la composición de la fase móvil, compuesta por acetonitrilo y HTBA 0,005M pH 5,0. Igualmente, y teniendo en cuenta las dos metodologías analíticas, cada una para la determinación separada de cada una de las sustancias, recibidas de la compañía Sumimed, se trabajaron diferentes sistemas de gradiente, con diversas composiciones tanto iniciales como finales, así como diferente perfil. Si bien se alcanzaron tiempos de retención apreciablemente menores y corridas aproximadamente 10 veces más cortas, los ensayos no se consideraron satisfactorios, observándose además asimetría en las señales cromatográficas.
La longitud de onda de la detección se estableció luego de la determinación del espectro de absorción molecular de las dos sustancias en una mezcla de acetonitrilo - HTBA, empleando concentraciones 32 y 80 mcg/mL, para piperacilina y tazobactam respectivamente. En cuanto a la temperatura de la fase estacionaria se realizaron ensayos que condujeron a una temperatura de 30°C como adecuada para la separación.
Algunos ensayos previos con concentraciones convenientes de las sustancias, permitieron iniciar los estudios de validación para la determinación conjunta de ellas en una sola corrida cromatográfica y por supuesto, con el mismo sistema cromatográfico.
Se trabajaron diferentes proporciones de acetonitrilo e hidróxido de tetrabutilamonio 0,005M de pH 5,0, siempre con elusión isocrática. Los ensayos, utilizando un flujo de fase móvil de 1 mL/min, se realizaron con proporciones de acetonitrilo situadas alrededor de 27%.
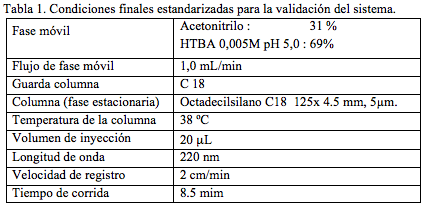
Según las experiencias y ensayos anteriores se establecieron las siguientes condiciones para proceder a la validación de la metodología (tabla 1).
.
Parámetros de idoneidad del sistema
Como paso previo al proceso de validación, se verificaron como parámetros de idoneidad del sistema cromatográfico, la repetibilidad, el factor de capacidad, el número de platos teóricos, el factor de asimetría y la resolución. Los valores hallados (tabla 2) satisfacen los requerimientos de la farmacopea americana [21]. La repetibilidad del sistema se evaluó mediante inyecciones sucesivas de una solución del estándar secundario de tazobactam en agua de concentración 80 mcg/mL y de manera separada, de piperacilina sódica en agua de concentración 640 mcg/mL. Como parámetro para la determinación se utilizaron los valores de área obtenidos.
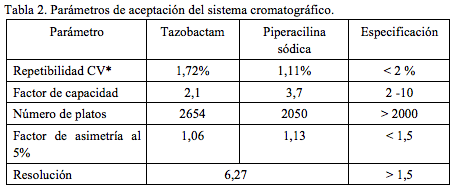
El valor de la resolución se estableció por análisis del cromatograma obtenido con la mezcla de las dos sustancias a las concentraciones ya mencionadas. El valor obtenido fue de 6,27, indicando así una separación completa y sin interferencias de cada una de las dos señales (Figura 1). Como tiempo muerto se tomó el tiempo de retención del solvente.
Validación de la metodología analítica
Selectividad
No se encontró que los componentes de la fase móvil generaran alguna señal que interfiera con las presentadas por la piperacilina o por el tazobactam, bajo las condiciones cromatográficas desarrolladas (figura 1).

La metodología recibida de la compañía Sumimed exige la selectividad del sistema cromatográfico frente a la ampicilina (producto de descomposición de la piperacilina). La señal presentada por la ampicilina se presenta a un tiempo de retención 1,20 minutos, la cual no interfiere con las observadas para los dos principios activos, piperacilina y tazobactam. La resolución entre las señales más cercanas, ampicilina y tazobactam, fue de 7,12. De acuerdo a la literatura [15, 16], el valor mínimo de resolución para una separación completa (100%) es de 1,50.
El cromatograma de la (Figura 1), ilustra la selectividad del sistema cromatográfico respecto de las dos sustancias objeto de análisis. El valor obtenido para la resolución fue de 7,38, garantizándose de esta forma separación completa de las dos señales, sin ningún tipo de interferencia.
En cuanto a los productos de descomposición, en la (Figura 2), se presenta el cromatograma obtenido con una solución de tazobactam que se guardó a 4 °C durante 16 días. Se observa una señal de un producto de descomposición a 2,10 minutos, la cual no interfiere con la debida al tazobactam. El valor de la resolución entre el tazobactam y el producto de descomposición fue de 3,01.
El cromatograma obtenido bajo las mismas condiciones con una solución de piperacilina presenta señales de baja intensidad a 1,27, 4,33, 6,62 y 10,58 minutos, las cuales no interfieren con la señal de la piperacilina, ni con la presentada por el tazobactam.
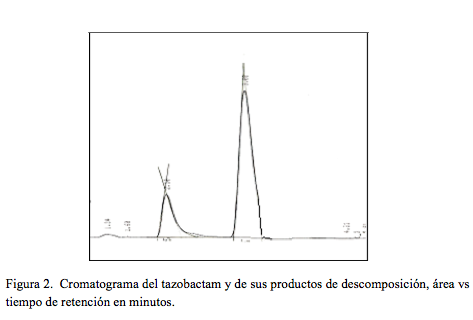
Linealidad
Para evaluar la linealidad del tazobactam se utilizaron concentraciones equivalentes a 20, 40, 60, 80 y 100 mcg/mL. En el caso de la piperacilina sódica se ensayaron concentraciones de 160, 320, 480, 640 y 800 mcg/mL. En ambos casos se realizaron tres ensayos de manera independiente (tres réplicas).
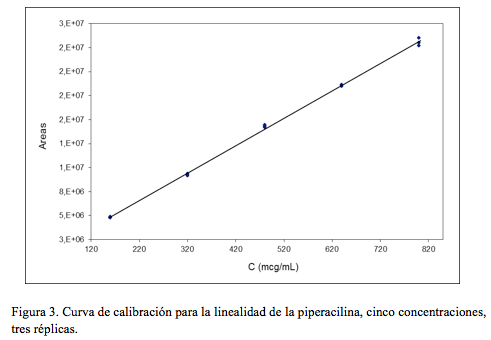
Como ecuaciones para las rectas de regresión, cinco concentraciones tres réplicas, se encontró:
y = 20101,01 x + 15365,76 r = 0,99919
y = 28633,97 x + 259409,86 r = 0,99937
para tazobactam y piperacilina, respectivamente.
En la (Tabla 3) se presentan los valores experimentales de los estadísticos t y F, para el análisis de intercepto, pendiente, regresión y falta de ajuste. Para las dos sustancias, de acuerdo al test de t de Student, se encuentran interceptos no significativos y pendientes significativas. Por otra parte, como los valores de F experimentales para la prueba F de Fisher son mayores que los valores de la tabla para la regresión y menores para el desvío, se encuentran regresiones significativas y desvíos (falta de ajuste) no significativos.
En la (Figura 3) se presenta la curva de calibración obtenida para el caso de la piperacilina.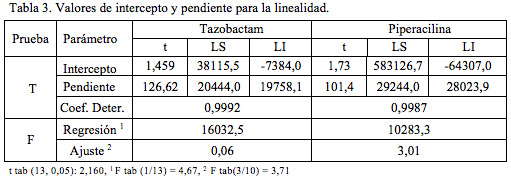
Precisión
La precisión del sistema para cada una de las dos sustancias, se verificó a nivel de repetibilidad para tres niveles de concentración y a nivel de precisión intermedia entre días.
Repetibilidad
Para evaluar la precisión a nivel de repetibilidad y para tres niveles de concentración, se realizaron cuatro determinaciones independientes para cada una de ellas (Tabla 4). Las concentraciones empleadas fueron 40, 60 y 80 mcg/mL para tazobactam y 320, 480 y 640 mcg/mL para piperacilina.
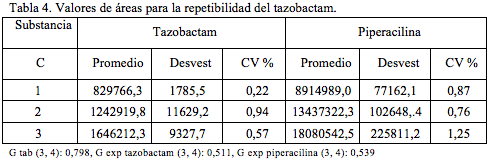
Como el valor de G experimental es menor que el valor tabulado para tres niveles de concentración, tres réplicas, se puede determinar un valor para la desviación estándar relativa (CV) global. Este valor hallado luego de ponderar los valores de CV para los tres niveles es 0,647% para tazobactam y 0,567% para la piperacilina, valores que satisfacen las especificaciones [15, 16], menor de 2%.
Precisión intermedia
La precisión intermedia entre días para el tazobactam se determinó utilizando una concentración de 60 mcg/mL y una concentración de 480 mcg/mL para la piperacilina, soluciones que se analizaron durante tres días, día 1, día 2 y día 4. En la Tabla 5 se presentan los resultados obtenidos. El valor promedio reportado corresponde al promedio de las áreas para 5 inyecciones.
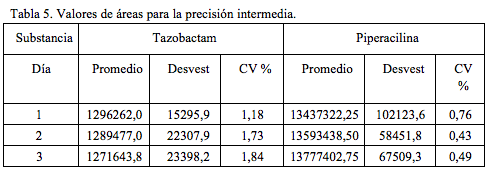
Se observa que, para cada sustancia y para cada uno de los tres días, el valor del coeficiente de variación es inferior a 2%, máximo valor permitido [14, 15].
Robustez
Para el estudio de robustez de la metodología, se tomó como referencia las recomendaciones de Youden y Steiner [22] adoptadas luego por Plackett y Burman [19] así como lo publicado por Vander y colaboradores [23].
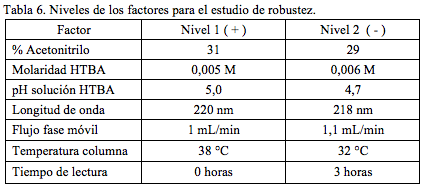
De acuerdo a lo establecido en la Ref. [23], se trabajó un diseño experimental en el cual se deben contemplar como mínimo 7 factores, que se evalúan mediante la realización de 8 experimentos independientes.
Para el estudio se utilizaron los reactivos y soluciones siguientes: acetonitrilo HPLC, hidróxido de tetrabutilamonio 0,005M pH 5,0, hidróxido de tetrabutilamonio 0,005M pH 4,7, hidróxido de tetrabutilamonio 0,006M pH 5,0, hidróxido de tetrabutilamonio 0,006M pH 4,7, solución de trabajo (tazobactan sódico 60 mcg/mL, piperacilina sódica 480 mcg/mL).
Como factores para el estudio se contemplaron los siguientes: porcentaje de acetonitrilo, concentración del hidróxido de tetrabutilamonio, pH de la solución de hidróxido de tetrabutilamonio, flujo de la fase móvil, temperatura de la columna, longitud de onda de la detección, tiempo de preparación de la solución de trabajo. En la Tabla 6 se presentan los niveles del estudio para cada factor real.
El orden de los factores se estableció de manera aleatoria, así como también el orden de los ensayos [21]. Para cada ensayo se realizaron entre 2 o 3 determinaciones, dependiendo de la variación observada. Para efectos de establecer la variación en los resultados por cambios en los factores que puedan presentarse durante el proceso de determinación simultánea de tazobactan sódico y de piperacilina sódica, se calculó para cada factor la diferencia entre la media de los valores de respuesta obtenidos para el nivel superior del efecto y la media de los valores de respuesta obtenidos para el nivel inferior del efecto. La diferencia se toma como un valor absoluto (Tabla 7). Como valor de referencia o de comparación para las diferencias se toma el valor que resulta de multiplicar la raíz de 2 por la desviación estándar obtenida en el estudio de precisión a nivel de repetibilidad y para la concentración equivalente al 100%. Estos valores corresponden a 11629,2 y 102648,0, para tazobactam y piperacilina respectivamente.
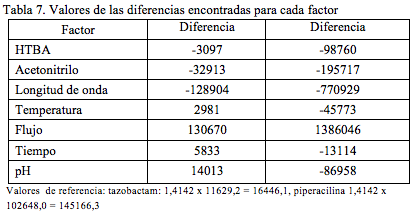
Teniendo en cuenta la coincidencia de los resultados tanto para el tazobactam como para la piperacilina, podemos concluir que la metodología es robusta y no se afecta por pequeños cambios en la concentración de hidróxido de tetrabutilamonio, temperatura de la columna, tiempo de análisis y pH. Por el contrario, la metodología es sensible frente a cambios en el porcentaje de acetonitrilo de la fase móvil, longitud de onda de la determinación y flujo de la fase móvil.
Aplicación
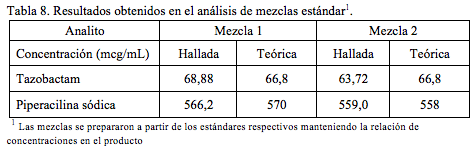
La metodología desarrollada y validada se aplicó para la valoración de una mezcla de tazobactam y de piperacilina sódica, elaborada por mezcla de soluciones de cada una de las dos sustancias, preparadas con los respectivos estándares secundarios, así como para la valoración de una mezcla de tazobactam sódico y de piperacilina sódica en una muestra conformada por tazobactam sódico y piperacilina sódica, materia prima para inyectables.
Se prepararon dos mezclas estándar de concentraciones 69,6 y 570 mcg/mL, y 66,8 y 558 mcg/mL para tazobactam y piperacilina respectivamente. En la Tabla 8 se reportan las cantidades de tazobactam y piperacilina sódica hallados experimentalmente aplicando la metodología validada, al igual que las cantidades teóricas de las mezclas.
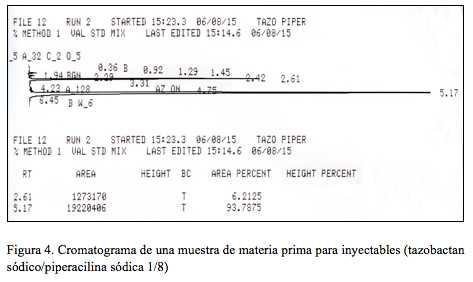
Para el caso de la materia prima inyectable, con una relación de concentraciones 1/8 de tazobactam y piperacilina, se prepararon 4 muestras, pesando 28,6, 31,1, 31,2 y 29,9 mg, pesos que se llevaron a 25 mL con agua para obtener concentraciones finales teóricas aproximadas de 66 y 530 mcg/mL para tazobactam y piperacilina respectivamente.
En la Figura 4 se presenta el cromatograma obtenido con la primera inyección de la segunda muestra.
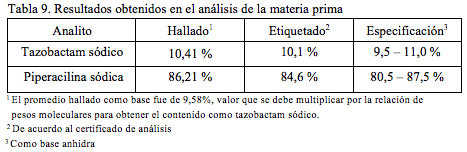
El promedio de los contenidos para cada una de las cuatro muestras de materia prima analizadas se indica en la Tabla 9.
CONCLUSIONES
Se estandarizó y validó una metodología analítica por HPLC en fase inversa para la determinación de tazobactan sódico y piperacilina sódica en materia prima destinada a la elaboración de inyectables. Durante la etapa de validación, la metodología demostró ser precisa, lineal, reproducible y selectiva frente a productos de descomposición obtenidos por almacenamiento a 4°C. Estas características la hacen adecuada y confiable para el objetivo propuesto. Además de ello, la metodología presentada es sencilla y se realiza en un tiempo adecuado respecto de otras encontradas en la literatura, a su vez que la fase móvil resulta menos agresiva con la fase estacionaria, optimizando su vida útil. . La metodología validada permite la determinación de las dos sustancias, individualmente o en mezcla, en inyectables de uso humano y puede ser igualmente utilizada para propósitos de identificación de las dos sustancias.
AGRADECIMIENTOS
A Laboratorios Sumimed, de Bogotá, Colombia, por su generoso apoyo para la realización del presente trabajo así como al Departamento de Farmacia de la Universidad Nacional de Colombia en cuyas instalaciones se llevó a cabo.
REFERENCIAS
1. D.M. Johnson, G.V. Doern, T.A. Haugen, J. Hindler, J.A. Washington, R.N. Jones, Comparative activity of twelve beta-lactam drugs tested against penicillin-resistant Streptococcus pneumoniae from five medical centers: Effects of serum protein and capsular material on potency and spectrum as measured by reference tests, Diagnostic Microbiology and Infections Disease, 25, 137 (1996). [ Links ]
2. I.K. Zaghloul, K. Nyda, A. Yacobi, The effect of tazobactam on the pharmacokinetics and the antibacterial activity of piperacillin in dogs, International Journal of Pharmaceutics, 153, 115 (1997). [ Links ]
3. T.R. Fritsche, H.S. Sader, R.N. Jones, Comparative activity and spectrum of broad-spectrum beta-lactams (cefepime, ceftazidime, ceftriaxone, piperacillin/tazobactam) tested against 12,295 staphylococci and streptococci: report from the SENTRY antimicrobial surveillance program (North America: 2001-2002), Diagnostic Microbiology and Infectious Disease, 47, 435 (2003). [ Links ]
4. H.S. Sader, A. Hsiung, T.R. Fritsche, N. Jones, Comparative activities of cefepime and piperacillin/tazobactam tested against a global collection of Escherichia coli and Klebsiella spp. with an ESBL phenotype, Diagnostic Microbiology and Infections Disease, 57, 341 (2007). [ Links ]
5. C.R. Bethel, A.M. Hujer, M.S. Helfand, R.A. Bonomo, Exploring the effectiveness of tazobactam against ceftazidime resistant Escherichia coli: insights from the comparison between susceptibility testing and beta-lactamase inhibition, FEMS Microbiology Letters, 234, 99 (2004). [ Links ]
6. E. Peyrin, Y. Guillaume, C. Guinchard, High-performance liquid chromatographic determination of tazobactam by precolumn derivatization, Journal of Chromatography B: Biomedical Sciences and Applications, 672, 160 (1995). [ Links ]
7. B. Tang, L. Ma, Y. Sun, H. Wang, Study on fluorescent property of degrading products of piperacillin and its analytical application, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 57, 217 (2001). [ Links ]
8. G. Pajchel, S. Tisky, Adaptation of capillary electrophoresis to piperacillin drug analysis, Journal of Chromatography A, 846, 223 (1999). [ Links ]
9. S.C. Marselos, H.A. Archontaki, Development and optimization of a reversed-phase high-performance liquid chromatographic method for the determination of piperacillin and tazobactam in tazocin injectable powder, Journal of Pharmaceutical and Biomedical Analysis, 29, 787 (2002). [ Links ]
10. V. Augey, P. Grosse, G. Albert, M. Audran, F. Bressolle, High-performance liquid chromatographic determination of tazobactam and piperacillin in human plasma and urine, Journal of Chromatography B: Biomedical Sciences and Applications, 682, 125 (1996). [ Links ]
11. A.P. Ocampo, K.D. Hoyt, N. Wadgaonkar, A.H. Carven, C.V. Puglisi, Determination of tazobactam and piperacillin in human plasma, serum, bile and urine by gradient elution reversed-phase high-performance liquid chromatography, Journal of Chromatography B: Biomedical Sciences and Applications, 496, 167 (1989). [ Links ]
12. T. Marunaka, M. Maniwa, E. Matsushima, Y. Minami, High-performance liquid chromatographic determination of a new beta-lactamase inhibitor and its metabolite in combination therapy with piperacillin in biological materials, Journal of Chromatography B: Biomedical Sciences and Applications, 431, 87 (1988). [ Links ]
13. European Pharmacopoeia, Council of Europe, 67075 Strasbourg Cedex, France, 4a. Ed., 2002, p. 1764-1766. [ Links ]
14. Farmacopea de los Estados Unidos de América, Formulario Nacional, USP 35 NF 30, The Pharmacopoeial Convention Inc., 2012, p. 4808. [ Links ]
15. Farmacopea de los Estados Unidos de América, Formulario Nacional, USP 35 NF 30, The Pharmacopoeial Convention Inc., 2012, p. 971. [ Links ]
16. FDA, CDER, "Guidance for Industry, Analytical Procedure and Validation", Agosto 2000. [ Links ]
17. ICH, "Validation of Analytical Procedures", Octubre de 1993. [ Links ]
18. LC-CG, "Validation of Analytical Methods: Review and Strategy", Febrero de 1998. [ Links ]
19. O.A. Quattrocchi, S.I. Abelaira y R.F. Laba, "Introducción a la HPLC, Teoría y Práctica", Buenos Aires, 1992. [ Links ]
20. "Guía de Validación de Métodos Analíticos", Colegio Nacional de Químicos Farmacéuticos Biólogos, México, 2011. [ Links ]
21. Farmacopea de los Estados Unidos de América, Formulario Nacional, USP 35 NF 30, The United States Pharmacopoeial Convention Inc., 2012, p. 285. [ Links ]
22. W.J. Youden, The Collaborative Test, Journal of the Association of Official Analitycal Chemists, 71, 195 (1990). [ Links ]
23. Y. Vander y otros, "Guidance for Robustness/Ruggedness", Universidad de Bruselas, Bélgica, 2003. [ Links ]
