










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
Print version ISSN 0034-7418
Rev. colomb. cienc. quim. farm. vol.43 no.2 Bogotá July/Dec. 2014
https://doi.org/10.15446/rcciquifa.v43n2.54209
Doi: http://dx.doi.org/10.15446/rcciquifa.v43n2.54209
Estudio comparativo de la calidad biofarmacéutica de marcas comerciales y multifuentes de tabletas de captopril y losartán del mercado colombiano
Comparative study of the biopharmaceutical quality of commercial and generic brands of captopril and losartan tablets in the Colombian market
Germán Eduardo Matiz Meloa
Erika Rodríguez Cavallob
a Afiliación inicial: Grupo de Investigación en Tecnología Farmacéutica, Cosmética y de Alimentos, Facultad de Ciencias Farmacéuticas, Universidad de Cartagena, Cartagena (Colombia).
Afiliación actual: Departamento de Farmacia, Universidad Nacional de Colombia, Cra. 30 No.45-03, Bogotá, D. C. (Colombia). Direcciones electrónicas: gmatizm@unicartagena.edu.co y gematizm@unal.edu.co
b Grupo de Investigación en Química Analítica y Biomedicina, Facultad de Ciencias Farmacéuticas, Universidad de Cartagena, Cartagena (Colombia). Dirección electrónica: erodriguezc1@unicartagena.edu.co
Recibido para evaluación: 14 de abril de 2014.
Aceptado para publicación: 21 de julio de 2014.
Resumen
La hipertensión arterial afecta a miles de personas en el mundo, y en Colombia permanece como una de las primeras causas de morbi-mortalidad. Los fármacos captopril y losartán constituyen la primera elección para su tratamiento. Con el fin de evaluar la conformidad de los productos y determinar su equivalencia biofarmacéutica, se evaluaron un total de 19 marcas comerciales disponibles en droguerías y farmacias de cuatro principales ciudades del país: Bogotá, Cartagena, Cali y Barranquilla. Para ello se evaluaron las características físicas, químicas y biofarmacéuticas de las tabletas, tales como variación de peso, dureza, desintegración, test de disolución, perfil de disolución, eficiencia de la disolución y valoración de principio activo, esta última, a partir de metodologías optimizadas y validadas. Los ensayos farmacopeicos se evaluaron segúún lo establecido en la USP 35. Los resultados permitieron establecer que todas las marcas analizadas cumplieron los criterios de aceptación establecidos en la farmacopea para cada principio y que el comportamiento biofarmacéutico de ellas era muy similar para ambos tipos de molécula. Los resultados de este trabajo permiten proponer a la comunidad científica la determinación de la equivalencia biofarmacéutica como elemento de apoyo en la toma de decisiones de compra en el servicio farmacéutico.
Palabras clave: captopril, losartán, biofarmacéuticos, disolución, factor de similitud f2.
Summary
Hypertension affects thousands of people around the world and in Colombia remains one of the leading causes of morbidity and mortality. The captopril and losartan drugs are the first choice for treatment. In order to assess the conformity of products and determine their biopharmaceutical equivalence, a total of 19 commercial brands available in drug stores and pharmacies in four major cities of the country (Bogotá, Cartagena, Cali and Barranquilla) were evaluated. To this end the physical, chemical and biopharmaceutical characteristics of the tablets, such as weight variation, hardness, disintegration, dissolution, dissolution profile, the dissolution efficiency and value of the active substance were evaluated. This latter from optimized and validated methodologies. Pharmacopeial assays were evaluated as provided in USP 35 NF 30. The results obtained allowed to establish that all models tested met the acceptance criteria in the Pharmacopoeia for each principle, and the biopharmaceutical behavior of brands is very similar for both types of molecule. The results of this work allows to propose the scientific community the determination of the biopharmaceutical equivalence as support in making purchasing decisions in the pharmaceutical service.
Key words: captopril, losartan, biopharmaceutical, dissolution, similarity factor f2.
Introducción
La hipertensión arterial (HTA) constituye uno de los principales factores de riesgo de enfermedades cardiovasculares. Afecta a mil millones de personas en el mundo y anualmente cobra la vida de nueve millones [1]. En Colombia, durante el bienio 2008-2010 las enfermedades cardiovasculares fueron responsables de cerca del 25% de la mortalidad general, constituyéndose en las primeras causas de muerte en el país. Un año después, el Sistema General de Salud clasificaba la HTA como la patología que generó tres veces más consultas médicas entre los colombianos y la principal causa de urgencias y hospitalizaciones [2].
Su reciente inclusión dentro del Plan de Acción Mundial 2013-2020, presentado por la OMS, busca trazar la ruta que permita a los países prevenirla y controlarla mediante estrategias fundamentales como el acceso a medicamentos de buena calidad, eficaces y de bajo coste [1]. En particular, el efecto de esta estrategia sobre el aspecto económico de los recursos del sector salud es evidente si se considera que en promedio la atención del paciente hipertenso cuesta el doble que la de un individuo normotenso [3].
En Colombia, los fármacos identificados como de primera elección en el tratamiento de la HTA son captopril y losartán. Ambos se encuentran en el listado de medicamentos esenciales del plan obligatorio de salud (POS) [4], y todas las entidades prestadoras de salud (EPS) las suministran según denominación multifuente (genérica). Lamentablemente la fuerte batalla comercial entre medicamentos innovadores y multifuentes, que ha existido desde hace ya varias décadas en el mercado farmacéutico nacional, ha creado un ambiente de incertidumbre entre médicos y pacientes, que se cuestionan su calidad e intercambiabilidad [5, 6]. Esta situación se agudiza tanto por la ausencia de una regulación adecuada y eficiente que vigile y controle su calidad, como por la falta de políticas claras de control de precios al comercio de medicamentos. El favorecimiento de la legislación nacional para la comercialización de multifuentes a fin de reducir los costos tanto para el sistema de salud como para el paciente no es suficiente para eliminar el ambiente de desconfianza que permanece en torno a ellos [7, 8], sobre todo por el hecho de encontrarse diferencias muy notables de precios, aun en productos multifuentes. Una alternativa que permite ofrecer elementos de juicio válidos para abordar este problema con datos experimentales consiste en desarrollar estudios biofarmacéuticos in vitro, cuya aplicación reúne evidencia importante sobre la calidad de los medicamentos con una mínima inversión.
A fin de determinar el nivel de cumplimiento y la equivalencia biofarmacéutica entre tabletas comerciales de captopril y losartán, en este trabajo se describen los estándares de conformidad oficiales (farmacopea americana-USP 35) [9] y no oficiales, tales como variación de peso, dureza, desintegración, test de disolución, perfil de disolución, eficiencia de la disolución y valoración de principio activo. Para esta última se empleó un método optimizado y validado que permitió separar las moléculas con mayor eficiencia cromatográfica y sensibilidad instrumental que la sugerida por la farmacopea.
Metodología
Reactivos y materiales
Metanol, acetonitrilo (grado HPLC), fosfato monobásico de sodio y fosfato dibásico de sodio (R. A.) empleados en la separación cromatográfica se obtuvieron de JT Baker (USA). Patrones secundarios de losartán (99,6%) y captopril (99,5%) fueron una amable donación de un laboratorio nacional de especialidades farmacéuticas. El agua empleada en la preparación de las disoluciones y como medio en los ensayos de disolución se purificó mediante un sistema Milli-Q (Millipore, Bedford, MA, USA). Todas las disoluciones preparadas se llevaron a baño de ultrasonidos (Cole-Parmer 8853), y las utilizadas para ensayos cromatográficos se filtraron en filtros de nailon de 0,45 µµm.
Instrumentación
Los ensayos de velocidad de disolución y perfiles de disolución se realizaron en un disolutor LID-6 (Vanguard Pharmaceutical Machinery, Inc., Spring, TX, USA), y la cuantificación del principio activo en las muestras se realizó en un espectrofotómetro UV-Vis S2100 (Cole Parmer, Vernon Hills, IL, USA). Un desintegrador VK-7 modelo 10-911-71-C (Vankel, Santa Clara, CA, USA) se usó en la determinación de la velocidad de desintegración. La dureza de las tabletas se evaluó con un durómetro Stokes-Monsanto (Warrington, PA, USA).
El pH de las disoluciones reguladoras se ajustó con un pH-metro OAKTON serie 510 (Vernon Hills, IL, USA). El sistema cromatográfico empleado en los ensayos de valoración de principio activo consistió en un cromatógrafo líquido LaChrom Elite (Hitachi, Tokyo, Japan) equipado con una bomba cuaternaria, desgasificador en línea, automuestreador, inyector automático y sistema de detección de arreglo de diodos (DAD).
Muestras
Se obtuvieron muestras (60 tabletas) de dos lotes diferentes de 8 marcas de captopril (50 mg) y 11 de losartán (50 mg) de diversos establecimientos farmacéuticos de las ciudades de Cartagena, Barranquilla, Bogotá y Cali. En ambos casos se incluyeron productos con denominación genérica, comercial y el innovador, observándose que estuviesen dentro de su período de vida útil y con registro sanitario vigente del Instituto Nacional de Vigilancia de Medicamentos y Alimentos (Invima). Con el fin de evitar el sesgo analítico, cada lote se identificó de modo aleatorio con letras de la A hasta la K, empleando los subíndices 1 y 2 para referirse a los dos lotes evaluados por marca. Durante los estudios, los productos se almacenaron en estanterías cerradas protegidas de la luz, a una temperatura de 25 °C y humedad relativa del 60%.
Ensayos físicos
Se desarrollaron según se describe en los correspondientes capítulos de la farmacopea americana 35 [9], con algunas adaptaciones.
Peso
(Capítulo 41, USP 35). Se pesaron de manera individual diez comprimidos de cada lote y marca, a los que previamente se les retiró el polvo de la superficie sin quebrarlos, y se registró su peso exacto. Este procedimiento se repitió seis veces por tableta y se calculó el valor medio y su desviación (RSD).
Dureza
(Capítulo 1217, USP 35). Una tableta de cada producto se colocó en el durómetro y se registró la fuerza necesaria para provocar su ruptura. Este procedimiento se realizó en seis tabletas de cada lote y marca y se determinó el valor promedio y RSD.
Desintegración
(Capítulo 701, USP 35). Una tableta de cada lote y marca se colocó en cada uno de los seis tubos de la canastilla del equipo de desintegración, empleando agua desionizada (37,0 +/- 1,0 °C) como líquido de inmersión. Se determinó el tiempo necesario para la completa desintegración de cada tableta y se calculó su valor medio y RSD.
Ensayos químicos
Test de disolución
(Capítulo 711, USP 35). Se evaluaron seis tabletas de losartán por lote y marca empleando el aparato II (paleta) del equipo, 900 mL de agua (37,0 +/- 0,5 °C) como medio para la disolución y velocidad de agitación de 50 rpm. Transcurridos 30 minutos del ensayo, una alícuota de 5 mL de disolución se extrajo de cada vaso, se filtró y se cuantificó por espectrofotometría (256 nm). No menos del 75 + 5% de la cantidad declarada en el marbete debe disolverse en 30 minutos (Q30, S1).
Entre tanto, las muestras de captopril requirieron el empleo del aparato I (canastilla) y 900 mL de disolución de ácido clorhídrico (0,01 N; 37,0 +/- 0,5 °C). La alícuota de 5 mL se sustrajo después de 20 minutos de ensayo y se cuantificó empleando una longitud de onda de 205 nm. No menos del 80 + 5% de la cantidad declarada en el marbete debe disolverse en 20 minutos (Q20, S1).
Perfil de disolución
Las condiciones experimentales y de cuantificación correspondieron a las descritas en el ensayo anterior (velocidad de disolución). Esta prueba, que no es oficial en la farmacopea americana, la recomienda la FDA como un ensayo muy indicativo del desempeño de los comprimidos [10]. Se realizó muestreándose de manera seriada cada vaso a los 1, 2, 3, 5, 7, 10, 15, 20 y 30 minutos para captopril y 1, 2, 5, 10, 15, 20, 30 y 45 minutos para losartán y calculándose el porcentaje de fármaco liberado (%) en cada punto, a lo largo del ensayo [11].
Eficiencia de la disolución
Es un parámetro utilizado para comparar los perfiles de disolución [12, 13]. Se define como el porcentaje de las áreas bajo la curva (ABC) descritas por los perfiles de disolución al ser trazados sobre planos cartesianos, respecto del valor matemático teórico de una disolución instantánea trazado en el mismo plano. Matemáticamente se expresa mediante la ecuación (1):
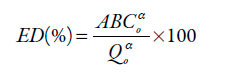 | [1] |
Donde ABC α o es la sumatoria de las áreas trapezoidales de la curva de disolución desde un tiempo cero hasta un tiempo α, que corresponde al tiempo del último punto experimental (150% del tiempo establecido en la monografía de la prueba de velocidad de disolución), y Q α o a se refiere al área del rectángulo delimitado por el porcentaje máximo de fármaco disuelto de manera instantánea y el tiempo α. Para efectos de comparación, se eligió la marca con mayor ABC, como la de referencia.
Valoración de principio activo
Para su evaluación, se optimizó y validó un método apoyado en HPLC-DAD para cada molécula. Las condiciones cromatográficas establecidas para el análisis de comprimidos de losartán incluyeron el uso de una columna Luna C8 (Phenomenex, endcapped, 150 ï´ 4,60 mm, 5 µm), empleando un programa de elución isocrática basado en el empleo de una mezcla tampón fosfato (18 mM, pH 6,8)/acetonitrilo (AcN), 85:15 %v/v, movido a flujo de 1,0 mL/min. La longitud de onda de detección fue 230 nm.
El método cromatográfico usado en los comprimidos de captopril se apoyó en el empleo de una columna Chromolith C18 (150 ï´ 4,6 mm), eluida en modo isocrático con una fase binaria (MeOH/H2O, 55:45 %v/v, 1 mL/min). El sistema de detección se ajustó a una longitud de onda de 220 nm.
Para la valoración del principio activo se determinó el peso promedio de seis tabletas de cada lote y marca, pulverizándolas a fin de preparar disoluciones de concentración 100 µg mL-1. Cada muestra se preparó por triplicado y cada disolución se inyectó tres veces en el equipo.
Análisis estadístico
Para todos los ensayos se calcularon las medias de los resultados entre los dos lotes de una misma marca mediante prueba t de student. p < 0,05 se consideraron significativas. Las diferencias entre las áreas bajo la curva (ABC) entre marcas se determinaron mediante análisis de varianza (Anova) y prueba de Tukey post hoc. p < 0,05 se consideraron significativas.
Resultados y discusión
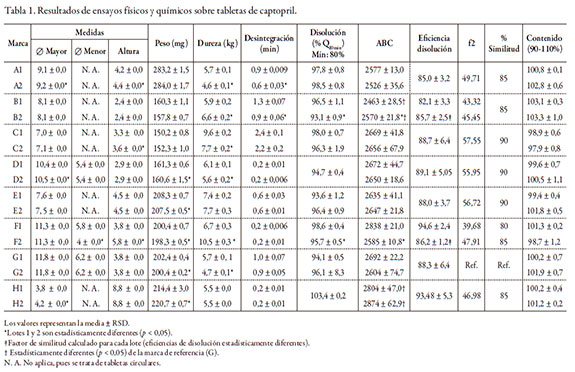
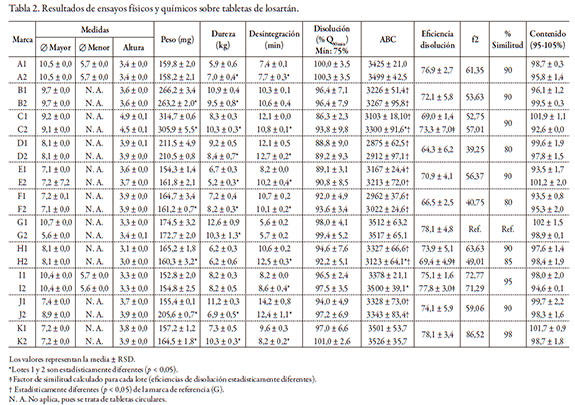
Los resultados de los ensayos físicos y químicos realizados sobre los diferentes productos sepresentan en las tablas 1 y 2 para captopril y losartán, respectivamente.
Ensayos físicos
La evaluación de las pruebas físicas realizadas en las muestras bajo estudio permitió identificar grandes diferencias entre los pesos de las diversas marcas. Los valores obtenidos oscilaron entre 150 y 284 mg para captopril y entre 152 y 314 mg para losartán. Las diferencias en las formulaciones, propias de cada laboratorio fabricante, y las dimensiones físicas de los productos, explican estas disimilitudes, sin que estén necesariamente relacionadas con variaciones en el contenido de principio activo o con fallas de desempeño en los ensayos de disolución. Entre lotes de la misma marca se detectaron variaciones de peso estadísticamente significativas en 5 de las 8 marcas de captopril y 6 de las 11 de losartán, pero por debajo del 2,0% en ambas medicaciones, concordante con la ausencia de registros de pesos individuales que difirieran significativamente del valor medio encontrado para el respectivo lote. Este comportamiento entre lotes permite inferir, en general, la ausencia de problemas de fabricación, mecánicos o de humedad excesiva durante la etapa de producción.
La dureza de las tabletas no se considera un elemento biofarmacéutico per se, sino más bien un elemento de control de calidad y reproducibilidad lote por lote. Los resultados obtenidos entre las diferentes marcas indicaron una fluctuación de la dureza de entre 5,2 y 8,7 kg cm-2 para captopril y de 6,0 a 11,5 kg cm-2 para losartán. Si bien estos datos tienen una diferencia significativa (RSD de 22% y 19%, respectivamente), las distintas tecnologías de producción que emplean las diferentes casas farmacéuticas bien explican las diferencias observadas entre marcas, sin que implique compromiso de la integridad del producto. Sin embargo, llama la atención que se encontraron diferencias en la dureza entre lotes en 6 de las 8 marcas de captopril (RSD entre 14% y 31%) y en 9 de las 11 de losartán (RSD entre 14% y 34%). No obstante, ninguna tableta presentó daño ni deterioro al ser retirada de su empaque primario, por lo que se puede inferir que existe una amplia tolerancia respecto de este ítem en el proceso de fabricación.
Los tiempos de desintegración obtenidos no superaron los 3 minutos para ninguna marca de captopril, ni 13 minutos para losartán, cumpliendo el criterio establecido por la USP para tabletas no recubiertas (< 30 minutos).
Ensayos químicos
Valoración de principio activo
La USP 35 ofrece una metodología para la valoración de losartán que implica el empleo de una fase móvil constituida por la mezcla H2O/AcN/ácido acético glacial, 59,8: 40,2: 0,4 %v/v, en la que se evidencia un alto contenido de componente orgánico y de concentración de iones hidronio. La tendencia actual de las metodologías analíticas se enfoca en desarrollar métodos amigables con el ambiente, lo que implica baja participación de disolventes orgánicos, amén de hacerlas más viables en función de costos. Por otra parte, el empleo de disolventes con valores de pH próximos a los extremos de la escala promueve el deterioro de la columna de separación cromatográfica, disminuyendo su eficiencia y acortando su vida útil. Considerando que el control de calidad es una herramienta fundamental que guía no solo la calidad de la materia prima empleada, sino también del proceso de fabricación y del producto terminado, es importante conseguir condiciones de separación cromatográfica que no promuevan su deterioro, que sean sostenibles y que generen el menor daño al ambiente.
Así, con estos elementos como objetivo, y buscando una adecuada sensibilidad, selectividad y transferencia de masa en el sistema cromatográfico, se llegó al empleo de una fase móvil constituida por la mezcla H3PO4 (18 mM, pH 6,8)/AcN, 85:15 %v/v, movida en elución isocrática.
En referencia a las dimensiones de la columna cromatográfica recomendadas por la USP para valorar los principios activos losartán y captopril (250 ï´ 4,6 mm), si bien sus dimensiones ofrecen mayor número de platos teóricos (N) para promover la separación cromatográfica de las sustancias de interés, también implican mayor tiempo de análisis y con ello mayor consumo de fase móvil durante la separación. De este modo, se optó por emplear columnas con menor longitud que permitieran obtener resultados analíticos en menor tiempo. La optimización de las condiciones de elución permitió alcanzar el mejor desempeño a partir de N para los principios estudiados.
La idoneidad cromatográfica obtenida para la separación de cada una de las sustancias de interés, medida en función de N, factor de asimetría y RSD entre áreas de réplicas de inyección, fue 834%, 2,03% y 0,52% para captopril y 936%, 1,09% y 0,43% para losartán, respectivamente. En ambos casos se cumplió la especificación exigida por la farmacopea en cuanto al factor de asimetría y RSD (< 2,0% y 2,0%, respectivamente).
El método optimizado se validó en función de linealidad, exactitud y precisión. Para propósitos de cuantificación durante la validación del método, se construyó una curva de calibrado con siete puntos obtenidos a partir de disoluciones patrón en el intervalo 25 a 1.000 µg mL-1 para losartán y 300 a 2.000 µg mL-1 para captopril. Se observó buena linealidad dentro del intervalo de concentraciones evaluadas para ambas moléculas (r > 0,999).
La exactitud se determinó preparando tres disoluciones patrón a los niveles de concentración 500, 800 y 1.000 µg mL-1 para cada molécula. El promedio de recuperaciones fluctuó entre 94,4% y 101% con RSD menores que 1,8% en todos los casos. Para determinar la repetibilidad y reproducibilidad del método, tres analistas prepararon en tres días diferentes una disolución patrón de cada molécula por triplicado al nivel 1.000 µg mL-1. El promedio de recuperaciones obtenidas en un mismo día osciló entre 96% y 97% (RSD < 0,42%), y entre días osciló entre 95% y 98% con valores de RSD inferiores a 0,3%. Estos datos confirman la buena reproducibilidad del método.
La aplicación del método optimizado y validado al análisis de las distintas marcas y lotes de losartán y captopril arrojó valores de recuperación de entre 92,6% y 103,2%, dependiendo de la molécula y RSD inferiores a 2,19%. De acuerdo con el criterio de aceptación de la USP 35 para losartán y captopril, el porcentaje presente en cada tableta debe oscilar entre un 95% y 105% y entre 90% y 110% de lo declarado en el marbete, respectivamente. Con apoyo en esta información, todas las muestras analizadas de captopril y losartán son conformes, a excepción de los lotes C2, E1 y F1 de losartán, y en los que la valoración de principio activo estuvo entre 92,6% y 93,5% (RSD < 1,9%) de lo declarado en el marbete.
Test de disolución
De acuerdo con los criterios establecidos por la USP para los fármacos en estudio, los resultados permitieron establecer que todas las marcas de ambas moléculas evaluadas cumplieron con holgura el criterio de aceptación S1 de la USP 35, ya que para captopril, la disolución mínima fue del 93,1%, cuando la norma exige 85%, y para losartán el guarismo inferior indicó un 86,3%, siendo exigible el 80% para el mencionado criterio.
Perfiles de disolución, área bajo la curva (ABC), eficiencia de la disolución (ED) y factor de similitud (f2)
A diferencia del test de disolución, en el que solo se evalúa la cantidad de fármaco liberado a un tiempo final, el perfil de disolución se construye analizando dicha liberación en múltiples tiempos a lo largo del ensayo, de modo que al final se define una curva que representa la forma en que ocurre la disolución durante todo el proceso. Para compararlos se calcularon los valores de las áreas bajo la curva (ABC) descritos por ellos [14, 15] y el factor de similitud f2, que compara punto por punto el proceso de disolución [16, 17]. Los valores promedio de disolución (%Q) por tiempo de muestreo se presentan en las tablas 3 y 4. Al comparar las distintas ABC y las ED (tablas 1 y 2), es evidente para captopril que la marca H tiene el más alto desempeño, pues su eficiencia sobrepasa el 93%; no obstante, no se toma esta marca como referencia, pues de hacerlo, todas las demás marcas parecerían deficientes, lo cual no refleja la realidad, pues no necesariamente una liberación tan inmediata tiene el mejor desempeño farmacocinético; por tanto, se optó por escoger la marca G como referencia. El análisis estadístico demuestra que las marcas A, C, D, E y F no difieren de la G en cuanto a la eficiencia de la disolución. La marca H difiere por encima, y la marca B es ligeramente inferior. Desde el punto de vista del factor de similitud f2, que es una prueba más potente, solo las marcas C, D y E equivalen a la marca G (diferencia < 10%); las demás difieren un 15%. Esto indica que, aun con pequeñas diferencias, todas las marcas de captopril se comportan in vitro de manera muy similar: en efecto, 6 de las 8 marcas habían liberado más del 80% del fármaco antes de los 7 minutos, y las otras 2 lo hicieron a los 10 minutos, cuando la norma (USP 35) exige ese porcentaje a los 20.
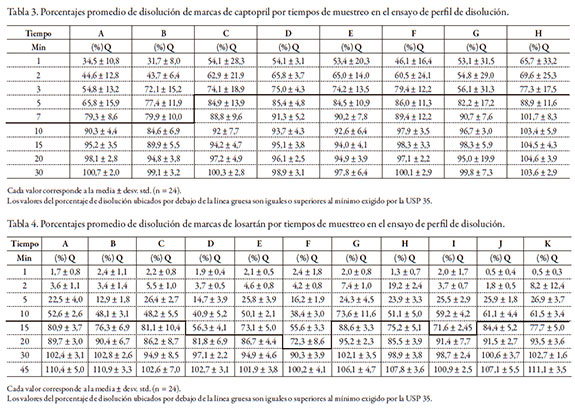
Para losartán, el comportamiento es un poco diferente. El análisis de las ABC revela que respecto de la marca de referencia (G) solo las marcas A, I y K (3 de 11) presentan comportamiento equivalente, y las ED oscilan entre el 66% y el 78%; no obstante, ninguna marca falló la prueba de disolución. La liberación del fármaco (75%) la lograron 7 de las 11 marcas antes de los 15 minutos, 3 a los 20 y solo una marca empleó los 30 minutos que reseña la monografía de la USP 35. Al revisar el factor f2, se encontró que 7 de las 11 marcas eran similares al de referencia (90%); para la marca H, uno de los lotes fue similar y el otro lo fue en un 85%, y dos marcas (D y F) lo fueron en un 80%.
En este trabajo también se quiso determinar si existía alguna correlación entre durezadesintegración-disolución de las diferentes marcas, que explicaran las diferencias de comportamiento durante la disolución. La prueba de desintegración fue la primera realmente biofarmacéutica que se hizo oficial en una farmacopea, la helvética (Suiza) en 1934, seguida por la farmacopea americana de 1950 (USP 14), pues según la ley de la disolución de Noyes-Whitney [18], la superficie de contacto entre soluto y solvente es crítica para la velocidad de la disolución, y era razonable pensar que una tableta que no se desintegraba no se disolvía con suficiente rapidez. Poco después, a finales de los años cincuenta, se descubrió que la desintegración por sí sola no era suficiente si los gránulos no se disolvían [19]. Por esta razón, en la farmacopea americana de 1970, la disolución aparece como prueba oficial de seis monografías. Los resultados encontrados en este trabajo se ajustan de manera correcta a lo dicho. Las figuras 1 y 2 correlacionan la dureza de la compresión con la desintegración y la eficiencia de la disolución. En ninguno de ambos casos -captopril y losartán- se observa tendencia alguna entre la dureza y las otras dos variables; es decir, a pesar de que hay marcas más duras que otras, los tiempos de desintegración no aumentan por ello. Solo la marca C de captopril, que es la más dura, alcanzó el mayor tiempo de desintegración, pero este no alcanzó los 3 minutos. La ED tampoco evidencia ninguna disminución respecto de las tabletas más duras. Lo que sí es evidente, y muy interesante, es que para ambos fármacos, tiempos más bajos de desintegración significaron tendencias de ED mayores, y viceversa, que no afectaron significativamente el comportamiento.
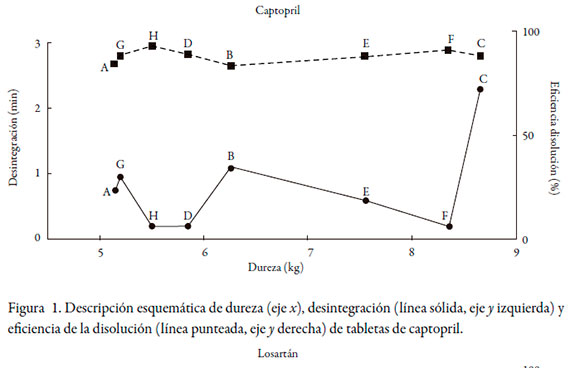
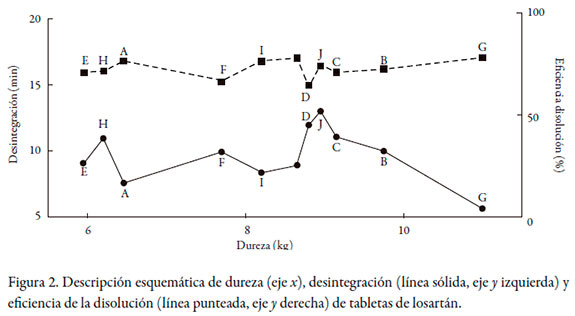
Tanto captopril como losartán pertenecen a la clase III del Sistema de Clasificación Biofarmacéutica (alta solubilidad, baja permeabilidad) [20-23]. De acuerdo con las directrices originales propuestas por Amidon et al. [24] y las revisadas en el trabajo colaborativo liderado por Polli et al. [25], fármacos de esta categoría pueden ser objeto de bioexención (exención de estudios de bioequivalencia), si muestran velocidades de disolución in vitro muy rápidas (85% en 15 minutos).
Todas las marcas de captopril analizadas y 7 de las 11 de losartán cumplieron esta condición, y por tanto podrían ser objeto de bioexención. No obstante, esta consideración aún es objeto de debate, por las múltiples variables que intervienen en el proceso de absorción (vaciamiento gástrico, grosor de las membranas, efecto de los alimentos, estado de hidratación, etc.), por lo que se propone que estas marcas de muy alta velocidad de disolución se consideren equivalentes biofarmacéuticos y que sin llegar a considerarlos intercambiables -connotación reservada solo para estudios de biodisponibilidad in vivo-, puedan tenerse como referencia para llevar a cabo la toma de decisiones, por ejemplo, desde el servicio farmacéutico de las entidades de salud como elemento de juicio para tomar la decisión de compra.
Conclusiones
Los resultados del trabajo permiten concluir que todos los productos evaluados cumplieron con las especificaciones de la farmacopea, y aunque tres lotes de losartán estaban bajos, no se puede concluir que el producto falló, pues no se debe ignorar que las condiciones de almacenamiento en el punto de venta final no siempre son las ideales. Además, es notable la similitud encontrada entre las marcas respecto del comportamiento de la disolución, y aunque esto no permite afirmar que son bioequivalentes, sí se puede afirmar que son equivalentes biofarmacéuticos, y esta comparación puede convertirse en un elemento de juicio decisorio por parte del servicio farmacéutico en el momento de elegir un proveedor.
Referencias
1. OMS. "Información general sobre la hipertensión 1. en el mundo". Ginebra (Suiza): Organización Mundial de la Salud, 2013. [ Links ]
2. ASIS. "Análisis de la situación de salud según regiones de Colombia". Bogotá (Colombia): Ministerio de Salud y Protección Social, 2013. [ Links ]
3. J.R. Chávez, C.G. Villafuerte, J.R. Legua, Tratamiento y costos farmacológicos de la hipertensión arterial no complicada en un hospital regional, Rev. Peruana Cardiol., 34(3), 181-190 (2008). [ Links ]
4. POS. Resolución 5521, Plan Obligatorio de 4. Salud. Bogotá (Colombia): Ministerio de Salud y Protección Social, 2013. [ Links ]
5. A. García, C. Hernández, C. Avendaño, Regulación de los medicamentos genéricos: evidencias y mitos, Inf. Terapéut. Sist. Nac. Salud, 34(3), 71-82 (2010). [ Links ]
6. O. Laosa, P. Guerra, J.L. López-Durán, B. Mosquera, J. Frías, Estudios de bioequivalencia: la necesidad de establecer la fiabilidad de los medicamentos genéricos, Rev. Peruana Med. Exp. Salud Públ., 26(4), 553-562 (2009). [ Links ]
7. J. Vásquez, K. Gómez, S. Rodríguez, Regulación en el mercado farmacéutico colombiano, Rev. Cienc. Soc. (Ven.), 16(2), 197-209 (2010). [ Links ]
8. V.J. Wirtz, M.R. Reich, M. Hernández-Ávila, Investigación para sustentar políticas farmacéuticas, Salud Publ. Mex., 50, S427-S428 (2008). [ Links ]
9. US Pharmacopeia, 35th Revision; The United States Pharmacopeial Convention. Rockville, MD, 2012. [ Links ]
10. FDA, Guidance for Industry: Dissolution testing of immediate-release solid oral dosage forms, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), 1997. [ Links ]
11. L.A. Franco, G.E. Matiz, I.B. Pájaro, Estudio biofarmacéutico comparativo de marcas comerciales de tabletas de ciprofloxacino disponibles en el mercado colombiano, Rev. Salud Publ. (Bogotá), 14(4), 695-709 (2012). [ Links ]
12. K. Khan, The concept of dissolution efficiency, J. Pharm. Pharmacol., 27(1), 48-49 (1975). [ Links ]
13. A. Cutrignelli, A. Trapani, A. Lopedota, M. Franco, D. Mandracchia, N. Denora et al., Griseofulvin/carrier blends: Application of partial least squares (PLS) regression analysis for estimating the factors affecting the dissolution efficiency, AAPS PharmSciTech, 12(4), 1019-1030 (2011). [ Links ]
14. T. O'hara, A. Dunne, J. Butler, J. Devane, A review of methods used to compare dissolution profile data, Pharm. Sci. Technol. Today, 1(5), 214-223 (1998). [ Links ]
15. N. Yuksel, A.E. Kan1k, T. Baykara, Comparison of in vitro dissolution profiles by ANOVA-based, model-dependent and-independent methods, Int. J. Pharm., 209(1), 57-67 (2000). [ Links ]
16. G. Frutos, J. Ocaña, Técnicas de remuestreo en la comparación de curvas de disolución de fármacos, An. Real Acad. Farm., 77(2), 1-19 (2011). [ Links ]
17. V.P. Shah, Y. Tsong, P. Sathe, J.-P. Liu, in vitro dissolution profile comparisonstatistics and analysis of the similarity factor, f2, Pharm. Res., 15(6), 889-896 (1998). [ Links ]
18. A.A. Noyes, W.R. Whitney, The rate of solution of solid substances in their own solutions, J. Am. Chem. Soc., 19(12), 930-934 (1897). [ Links ]
19. A. Dokoumetzidis, P. Macheras, A century of dissolution research: From Noyes and Whitney to the biopharmaceutics classification system, Int. J. Pharm., 321(1), 1-11 (2006). [ Links ]
20. M. Lindenberg, S. Kopp, J.B. Dressman, Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system, Eur. J. Pharm. Biopharm., 58(2), 265-278 (2004). [ Links ]
21. N.A. Kasim, M. Whitehouse, C. Ramachandran, M. Bermejo, H. Lennernäs, A.S. Hussain et al., Molecular properties of WHO essential drugs and provisional biopharmaceutical classification, Mol. Pharm., 1(1), 85-96 (2004). [ Links ]
22. N.K. Sachan, A. Bhattacharya, S. Pushkar, A. Mishra, Biopharmaceutical classification system: A strategic tool for oral drug delivery technology, Asian J. Pharmaceut., 3(2), 76-81 (2009). [ Links ]
23. T. Takagi, C. Ramachandran, M. Bermejo, S. Yamashita, L.X. Yu, G.L. Amidon, A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan, Mol. Pharm., 3(6), 631-643 (2006). [ Links ]
24. G.L. Amidon, H. Lennernäs, V.P. Shah, J.R. Crison, A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability, Pharm. Res., 12(3), 413-420 (1995). [ Links ]
25. J.E. Polli, L.X. Yu, J.A. Cook, G.L. Amidon, R.T. Borchardt, B.A. Burnside et al., Summary workshop report: Biopharmaceutics classification system. Implementation challenges and extension opportunities, J. Pharm. Sci., 93(6), 1375-1381 (2004). [ Links ]
