










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Químico - Farmacéuticas
Print version ISSN 0034-7418
Rev. colomb. cienc. quim. farm. vol.44 no.2 Bogotá May/Aug. 2015
https://doi.org/10.15446/rcciquifa.v44n2.56291
Molecular docking study of naturallyoccurring compounds as inhibitors of N-myristoyl transferase towards antifungal agents discovery
Acoplamiento molecular de compuestos de origen natural como inhibidores de la N-miristoil transferasa en el descubrimiento de nuevos agentes antifúngicos
Camilo Guerrero-Perilla, Freddy A. Bernal, Ericsson D. Coy-Barrera *
Laboratorio de Química Bioorgánica, InQuiBio, Departamento de Química, Facultad de Ciencias Básicas y Aplicadas, Universidad Militar Nueva Granada, Cundinamarca, Colombia. A. A.: 49300. Fax: +57 1 2147280.
* Correo electrónico: ericsson.coy@unimilitar.edu.co.
Recibido para evaluación: 20 de enero de 2015.
Aceptado para publicación: 5 de mayo de 2015.
Summary
Fungal infections currently remain as a common problem in public health. Actually, drug discovery programs are oriented to the searching for lead structures. Virtual screening and molecular docking constitute great alternatives in order to find hit compounds. Novel infection targets can also be defined and employed together with molecular docking tools in drug discovery programs. Thus, thirty-two natural compounds were docked within the active site of N-myristoyl transferase (NMT) as antifungal enzyme target. From tested compounds, alkaloids, flavonoids, xanthones, and quinones exhibited strongest mean interaction with NMT than terpenoids, coumarins and phenolics. Particularly, affinities for one aporphine alkaloid, a prenylated flavonoid and two xanthones resulted to be comparable with that of previously reported synthetic inhibitor. Several hydrophobic and polar contacts were demonstrated by comparing different computational tools. The present results let to establish three possible lead structures to develop antifungal drugs although subsequent SAR analyses are still required.
Key words: Molecular docking, natural products, antifungal, N-myristoyl transferase.
Resumen
Las infecciones causadas por hongos continúan siendo un problema de salud pública en la actualidad. De hecho, existen diversos programas para el descubrimiento de fármacos enfocados en la búsqueda de estructuras plantilla. El mapeo virtual junto con docking molecular constituye una alternativa importante para encontrar potenciales estructuras promisorias. Mediante herramientas de docking molecular se pueden definir nuevos blancos terapéuticos para combatir diversas infecciones. Por tanto, se llevó a cabo el estudio del acoplamiento molecular a treinta y dos compuestos de origen natural, empleando la N-miristoil transferasa (NMT) como blanco enzimático antifúngico. De los compuestos ensayados, alcaloides, flavonoides, xantonas y quinonas mostraron interacción media más fuerte con la NMT que los terpenos, cumarinas y fenólicos. Particularmente, la afinidad encontrada para un alcaloide aporfínico, un flavonoide prenilado y dos xantonas resultó comparable con la encontrada para un inhibidor sintético reportado. En el presente trabajo se demostraron varias interacciones tanto hidrofóbicas como hidrofílicas mediante diversas herramientas computacionales. Los resultados encontrados permiten establecer tres posibles estructuras promisorias para el desarrollo de fármacos antifúngicos, aunque se requiere aún de estudios de relación estructura-actividad.
Palabras clave: docking molecular, productos naturales, antifúngicos, N-miristoil transferasa.
Introduction
So far, fungal infections remain being a public health problem. The use of antimicrobial drugs always carries the risk that resistance appears due to adaptable nature of the microorganism populations [1]. At this regard, continuous searching for antifungal agents on new targets or new action mechanisms is still required. Systemic antifungal agents can be generally grouped on basis of their action mechanism in pathogenic fungi, mainly in cell membrane, cell wall and intracellular action [2]. In depth, eight targets have been defined for antifungal therapy as follows: fungal ergosterol synthesis inhibitors, squalene epoxidase inhibitors, ergosterol disruptors, glucan synthesis inhibitors, chitin synthesis inhibitors, nucleic acid synthesis inhibitors, protein synthesis inhibitors and microtubules synthesis inhibitors [3].
N-Myristoyl transferase (NMT) catalyzes the transfer of the 14-carbon saturated fatty acid myristate from myristoyl-CoA to the N-terminal glycine residue of a variety of eukaryotic cellular and viral proteins [4-6]. NMT's are involved in a wide variety of biological processes and include protein kinases, kinase substrates, protein phosphatases, α-subunits of many heterotrimeric G-proteins and endothelial cell nitric oxide synthase [6]. NMT participates in diverse biological processes, including signal transduction cascades and apoptosis [4, 5]. NMT has even been employed as drug target for human pathogens as Candida and proposed as drug target of antitrypanosomatid and antimalarial therapeutics [7]. NMT has been reported as a potential drug target since it is involved in signaling networks and it is essential for the growth of human pathogens such as C. albicans [8]. The accepted action mechanism of NMT is schematized in Figure 1a [9, 10].
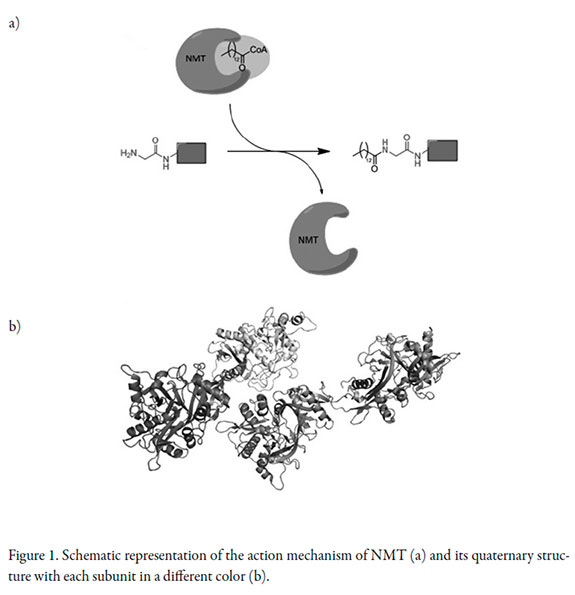
There are several chemical structure types which have demonstrated antifungal activity such as azoles, morpholines and other heterocyclic compounds [3]. In the same way, different natural compounds have been reported as possible antifungal agents, including phenols, flavonoids, coumarins, quinones, saponins, xanthones, alkaloids and terpenoids [11-13]. However, the screening of a great number of compounds is a limiting step in current researches. Virtual screening is a powerful computational tool that let to accomplish a preliminary searching of active compounds by means of molecular data banks [10]. Molecular docking consists of accurately prediction of the structure of a ligand within the constraints of a receptor binding site and to correctly estimation of the binding strength [14, 15]. Although docking scores are highly susceptible to preparation conditions of active sites and ligands, the comparison of them under exactly determined conditions constitutes a great indicative of the relative activity of the tested molecules as inhibitors of the target enzyme [14-18]. Molecular docking has increased its use as a fundamental tool of research on drug discovery and has been then improved in recent years [16-18].
The aim of the present research was to perform a molecular docking study of thirtytwo naturally-occurring compounds against N-myristoyl transferase from Candida albicans. The docked natural compounds were previously reported as possible antifungal leads. Here NMT is proposed as an important antifungal agent target. The results are also analyzed by means of pharmacophore modeling.
Methodology
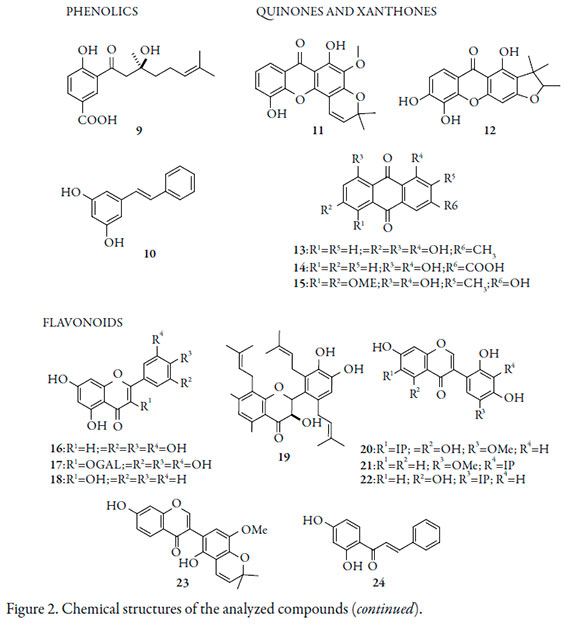
Thirty-two natural compounds were selected as common reported antifungal natural compounds from literature [11, 13]. All chemical structures were sketched in ChemBioDraw Ultra 12.0 (CambrigdeSoft) and shown in Figure 1. Conformational searching was carried out in Spartan'14 (Wavefunction, Inc.) employing AM1 semiempirical method. Geometry optimization of the lowest energy conformer was accomplished by density functional methods (DFT) at the B3LYP/6-31G* level. The same procedure was repeated with the structure of 33, a reported synthetic heterocyclic inhibitor (Figure 1).
NMT pdb file was downloaded from Protein Data Bank (PDB) with the code 1IYL (Figure 2). The structure of the protein was submitted for removing of hydrogen atoms and crystalized ligand structures. The active site was delimited based on reported residues at PDB and complemented with searching of close residues until 5 Å far crystallized inhibitor supported by AutoDock Tools (The Scripps Research Institute). Thus, active site was constituted by following residues: Asp-110, Phe-117, Tyr-225, Leu-337, Tyr-354, Asn-392, Cys-393, Leu-394, and Leu-451. All molecular docking assays were carried out using flexible residues.
Ligand and protein was docked employing AutoDock/Vina [19]. This program is based on the Iterated Local Search global optimizer by means of which several steps of mutation and local optimization are performed and then accepted by Metropolis criterion [19]. The strongest docked pose was analyzed in Pymol (Schödinger). All calculations were performed in a Dual Intel Xeon® processor CPU @ 2.6 GHz of Intel system origin, with 16 GB DDR3 RAM. Ligand preparation processes was run under Microsoft Windows 8 operating system while docking was compiled under Ubuntu 12.04 operating system.
Residual interactions maps were obtained with Discovery Studio (Accelrys software Inc.) using the AutoDock/Vina outputs (pdbqt files) for compounds that exhibited highest affinity energy. Pharmacophore analysis was also performed for those compounds. This analysis was accomplished with pharmacophore modeling tool from LigandScout (Gerhard Wolber and Inte:Ligand GmbH). Suitable pdb files from Discovery Studio were used as input in LigandScout.
Results and discussion
Performance of the AutoDock Vina algorithm on NMT - ligand complexation was evaluated by re-docking protocol applied on co-crystallized structure of 33. Ten runs for 33 were carried out and root mean square deviation (RMSD) values were then calculated. A mean RMSD of 2.08 Å (±0.45 Å) was found. This RMSD value was accepted as validation criteria for the docking algorithm. Moreover, some difference between co-crystallized and calculated pose can be expected due to the freedom degrees of the structure of 33. The calculated pose was also validated by observing molecular contacts with NMT pocket residues and comparing them with those reported in the crystal structure of the NMT-33 complex.
Thirty-two compounds previously reported as antifungal substances were selected as possible lead structures (Figure 2). These were submitted to conformational searching and the most stable conformer was then optimized at DFT level. Inhibitory potential of the obtained structures to NMT were evaluated by means of molecular docking. The results of molecular docking were first analyzed in terms of affinity energy (Table 1).
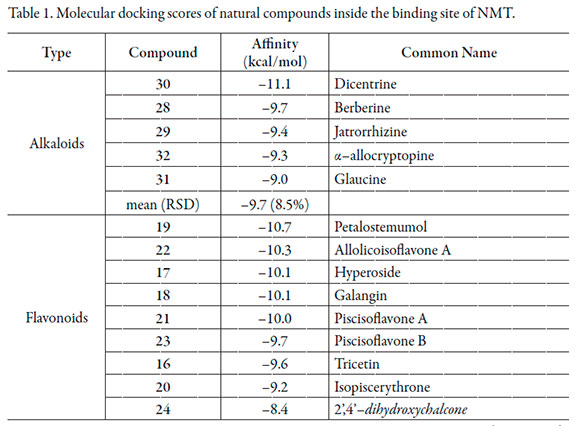
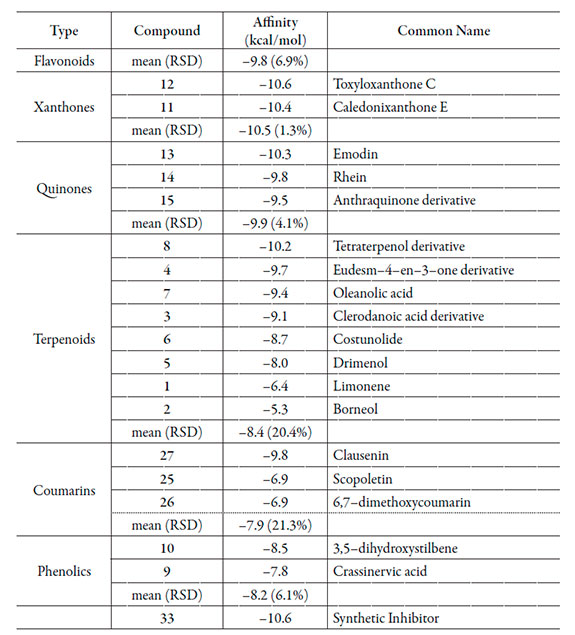
Lowest Affinity with the NMT-active site (mean affinity between -7.9 and -8.4 kcal/mol and high RSD values) were found for coumarins, terpenoids and phenolics (Table 1). Quinones, flavonoids and alkaloids resulted to be very similar regarding mean affinity from analyzed compounds with lower RSD values. Comparable antifungal effect could be expected due to NMT inhibition. Xanthones were those with the highest mean affinity energy toward NMT, and let us to establish them as ideal lead compound candidates. Moreover, these structures (11 and 12) exhibited very close affinity values to that of the synthetic inhibitor 33 despite the great chemical structural differences. In depth, the above mentioned xanthones are quite structurally dissimilar (11 possess a pyran-ring while 12 possess a furan-ring). However, both compounds are 1,3,5-trioxygenated. Simultaneously, pyran and furan rings seem to be not related to NMT interaction according to the found affinity. Further SAR studies for 1,3,5-trioxygenated xanthones are therefore required.
On the other hand, NMT-flavonoids interaction values were found to be highly variable (ranging between -8.4 to -10.7 kcal/mol; RSD = 8.4%) demonstrating that affinity depends on structure. On comparing 16, 17 and 18, structure-affinity relationships can be observed; 17 and 18 affinities exhibited identical value, indicating no effect by the presence of galactose moiety at C3-O. Nevertheless, oxygenation at C3 resulted to be essential to the interaction with NMT-active site (significant difference between 16 and 17 or 18 docking scores). Regarding isoflavones 20-22, affinity demonstrated to be very sensible to the positions of isoprenyl and hydroxyl group. The interaction 19-NMT was significantly higher to those of other docked flavonoids. However, other prenylated flavonols were not tested in the present study, so clear conclusions can't be established.
Ligand-enzyme affinity for alkaloids was found to be low (-9.7 kcal/mol as mean docking score) excepting 30, which demonstrated to achieve an important strong affinity with NMT (-11.1 kcal/mol; Table 1). The presence of a methylenedioxy group instead two methyl groups was preferred for the interaction with NMT (on comparing 30 with 31). 33 corresponds to a synthetic heterocyclic compound without structural similarity from 30. This fact let us to expect a different action mode for the above mentioned compound despite the closely related affinity energy. Compound 30 do not showed any polar contact with the amino acid residues from the NMT-active site, indicating a purely hydrophobic interaction with it. Moreover, 3D enzyme-ligand complex structure (Figure 3a) demonstrated a coplanar location of 30 with respect to Tyr-225 inferring a strong π- π interaction between phenyl rings (~3.6 Å).

No polar contacts were detected for compound 19. Instead of these the isoprenyl group at C6 on B ring was located between phenyl ring of Tyr-225 and Asn-392 amino group (3.8 and 3.7 Å, respectively) (Figure 3b). Other possible hydrophobic interaction between Tyr-354 and its B ring could be proposed, although this is not a pure π-π interaction due to the lack of coplanarity.
Two hydrogen bondings between amino group of Asn-392 and 12 were found (Figure 3c). An oxygen atom of the dibenzo-γ-pyrone moiety and a hydroxyl group at C5 were responsible of these polar interactions (2.3 and 2.6 Å, respectively). Simultaneously, a near π-π hydrophobic interaction can be inferred among Tyr-225 and the heterocyclic ring of the dibenzo-γ-pyrone moiety (mean distance = 3.6 Å). The same polar contacts were found for 11 (1.9 and 2.6 Å; Figure 3d). Moreover, hydrogen bonding between hydroxyl groups at Tyr-225 and C2 of 11 was clearly evident (2.2 Å). A weak hydrophobic interaction between A ring and Tyr-225 (5 Å aprox.) and a direct π - π interaction between B ring and Phe-240 (mean distance of 3.6 Å) were also identified in the NMT-11 complex. In spite of the differences at molecular level and the found interactions in 11 and 12, not significant differences on affinity energy were established (0.2 kcal/mol, which can be attributed to the difference inherent to the molecular docking method). Therefore, 5-hydroxy-dibenzo-γ-pyrone structure could be subjected to further QSAR studies.
In order to improve the information extracted from molecular docking results, docked molecules were analyzed regarding residual interactions with Discovery Studio (Accelrys software Inc.). Residual interactions map (Figure 4) highlights additional enzyme- ligand interactions that are not evident from the docked results viewed with Pymol. Here, only hydrophobic interactions between 30 and NMT were defined (Figure 4a).
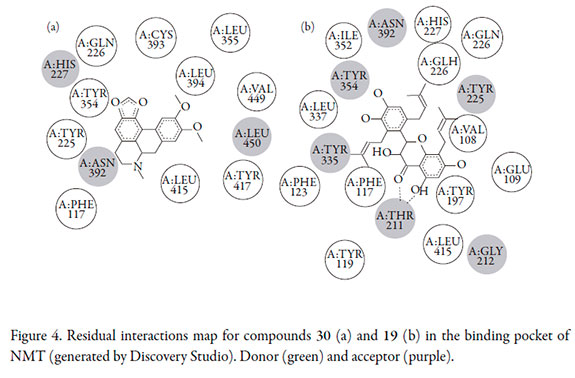
In this enzyme-ligand complex, π-electron acceptor character from His-227, Asn-392 and Leu-450 were showed. A double hydrogen bonding between Thr-211 residue and the carbonyl and hydroxyl groups at γ-pyrone moiety was observed for compound 19. These H-bonds were not evident in the primary docked structure since Thr-211 residue was not considered into the set of residues comprising the active site of the NMT. The present analysis can determine some interaction from ortho-dihydroxy group of B ring in 19 with Tyr-354 residue as H-acceptor. Accordingly, Val-108, Tyr-107, Phe-117, and Phe-339 demonstrated to be π- and H-donors to 19 interacting thus with different parts of the molecule (Figure 4b).
Finally, pharmacophore analyses for 11, 12, 19 and 30 were developed (Figure 5) employing the pharmacophore modeling tool from LigandScout (Gerhard Wolber and Inte:Ligand GmbH). π-π interaction between aromatic ring at quinoline system of 30 and Tyr-225 residue was confirmed as the main interaction in the minimum energy pose for the resulting ligand-enzyme complex (Figure 5a). The importance of isoprenyl groups as the key hydrophobic regions for the interaction with NMT-pocket residues was established by pharmacophore modeling (Figure 5b). This fact could be inferred from the differential affinity energy of 19 from the rest of the tested flavonoids (Table 1). On the other hand, LigandScout was not able to define interaction between 19 and Thr-211 residue in spite of the spatial proximity between them (Discovery Studio showed this interaction as mentioned above and observed in Figure 4b).
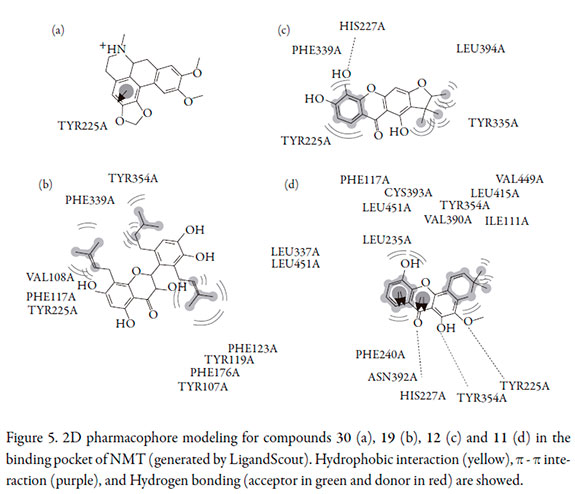
For compound 11, several structural requirements to interact with NMT under pharmacophore modeling were found (Figures 5d). Hydrophobic contacts from B ring and methyl groups on pyrone moiety were evident. Moreover, hydrogen bonds for carbonyl, hydroxyl and methoxy groups were confirmed by interacting with Ans-392, Tyr-354, Tyr-225 residues, respectively. Finally, π-π interactions among A ring of the γ-pyrone moiety and near aromatic residues were established as shown in Figure 5d. For compound 12, hydrophobic contacts for B ring and methyl groups from the furan ring could be proposed as key interactions (Figure 5c). Furthermore, hydrogen bonding between His-227 and one hydroxyl group in B ring of 12 can be proposed from the pharmacophore modeling (Figure 5c). Significant differences with regard to the best docking poses and the corresponding ligand-pocket residues interactions were found. Therefore, completely different action mechanisms on NMT inhibition should be expected for the tested and described natural compounds.
Indeed, compounds 30 and 19 have been reported as antifungal and/or antimicrobial agents. 30 was reported as the most active alkaloid from Glaucium oxylobum compared with glaucine, protopine, α-allocryptopine, and O-methylflavinantine [20]. In this study, dicentrine (30) resulted to be highly active against Microsporum gypseum, Trichophyton mentagrophytes and Epidermophyton floccosum (α-allocryptopine was only comparable against Epidermophyton floccosum). In a similar way, 19 was reported as the most active compound from a set of seven structurally related flavonoids (four times more active than others) [21]. This marked activity was demonstrated against Candida albicans, Cryptococcus neoformans, Mycobacterium intracellulare, Escherichia coli, Staphylococcus aureus and Bacillus subtilus. Compounds 12 and 11 have also been reported as potential antifungal natural compounds [22, 23]. In these literature reports, 12 showed markedly antifungal activity against Candida albicans [22], while 11 exhibited the strongest activity against Aspergillus fumigatus [23]. However, Larcher et al. [23] suggested a chitin synthesis inhibition mechanism. Furthermore, 12 was reported as inhibitory substance of B. subtilis and S. aureus with medium activity respect another natural xanthones from Cudrania cochinchinensis [24]. Antifungal activity of natural and synthetic xanthones have been analyzed and reported against several fungal strains [25, 26], however there are not previous QSAR analyses.
In conclusion, thirty-two compounds from natural sources were tested as inhibitors of the N-myristoyl transferase via molecular docking. From docked compounds, xanthones demonstrated to be able to form strong complexes with NMT. However, a prenylated flavonoid and an aporphine alkaloid were found as those with strongest interaction with NMT. The present findings let to propose at least four compounds as possible lead structures as potent inhibitors of NMT. At the same time, NMT could constitute an efficient and unexplored target for in-silico antifungal drug discovery researches. Nevertheless, due to the well-known limitations of the molecular docking calculations, future studies must be conducted using molecular dynamics simulations in order to get more reliable data as well as the validation at in-vitro level of these calculations in further binding affinity experiments with the aim to perform QSAR studies on compound series from the detected hits as second step in our research.
Acknowledgments
Authors thank Vicerrectoría de Investigaciones at Universidad Militar Nueva Granada - Validity 2013 for the financial support of the Project CIAS-1331.
References
1. J.B. Anderson, Evolution of antifungal - drug resistance: Mechanisms and pathogen fitness, Nature Reviews, 3, 547 (2005). [ Links ]
2. R.E. Lewis, Current concepts in antifungal pharmacology, Mayo Clinic Proceedings, 86, 805 (2011). [ Links ]
3. M.K. Kathiravan, A.B. Salake, A.S. Chothe, P.B. Dudhe, R.P. Watode, M.S. Mukta, S. Gadhwe, The biology and chemistry of antifungal agents: A review, Bioorganic & Medicinal Chemistry, 20, 5678 (2012). [ Links ]
4. J.A. Boutin, Myristoylation, Cellular signaling, 9, 15 (1997). [ Links ]
5. S. Sogabe, M. Masubuchi, K. Sakata, T.A. Fukami, K. Morikami, Y. Shiratori, H. Ebiike, K. Kawasaki, Y. Aoki, N. Shimma, A. D'Arcy, F.K. Winkler, D.W. Banner, T. Ohtsuka, Crystal structures of Candida albicans N-myristoyltransferase with two distinct inhibitors, Chemistry & Biology, 9, 1119 (2002). [ Links ]
6. R.S. Bhatnagar, K. Fütterer, T.A. Farazi, S. Korolev, C.L. Murray, E. Jackson-Machelski, G.W. Gokel, J.I. Gordon, G. Waksman, Structure of N-myristoyltransferase with bound myristoylCoA and peptide substrate analogs, Nature, 5, 1091 (1998). [ Links ]
7. M.H. Gelb, W.C. Van Voorhis, F.S. Buckner, K. Yokoyama, R. Eastman, E.P. Carpenter, C. Panethymitaki, K.A. Brown, D.F. Smith, Protein farnesyl and N-myristoyl transferases: Piggy-back medicinal chemistry targets for the development of antitrypanosomatid and antimalarial therapeutics, Molecular & Biochemical Parasitology, 126, 155 (2003). [ Links ]
8. M.H. Wright, W.P. Heal, D.J. Mann, E.W. Tate, Protein myristoylation in health and disease, Journal of Chemical Biology, 3, 19 (2010). [ Links ]
9. E. Thinon, R.A. Serwa, M. Broncel, J.A. Brannigan, U. Brassat, M.H. Wright, W.P. Heal, A.J. Wilkinson, D.J. Mann, E.W. Tate, Global profiling of co-and post-translationally N-myristoylated proteomes in human cells, Nature Communications, 5, 4919 (2014). [ Links ]
10. D. Rognan, Virtual screening by molecular docking. In: "Chemogenomics and chemical genetics: A user's introduction for biologists, chemists and informaticians," E. Maréchal et al. (editors), Springer-Verlag Berlin Heidelberg, 2011. [ Links ]
11. T. Arif, J.D. Bhosale, N. Kumar, T.K. Mandal, R.S. Bendre, G.S. Lavekar and R. Dabur, Natural products - antifungal agents derived from plants, Journal of Asian Natural Products Research, 11, 621 (2009). [ Links ]
12. M.J. Abad, M. Ansuategui, P. Bermejo, Active antifungal substances from natural sources, ARKIVOC, vii, 116 (2007). [ Links ]
13. T. Arif, T.K. Mandal, R. Dabur, "Natural products: Anti-fungal agents from plants". In: Opportunity, Challenge and Scope of Natural Products in Medicinal Chemistry, V.K. Tiwari, B.B. Mishra (editors), Research Signpost, Kerala, India, 2011, pp. 283-311. [ Links ]
14. D.B. Kitchen, H. Decornez, J.R. Furr, J. Bajorath, Docking and scoring in virtual screening for drug discovery: Methods and applications, Nature Reviews, 3, 935 (2004). [ Links ]
15. E. Yuriev, P.A. Ramsland, Latest developments in molecular docking: 2010-2011, Journal of Molecular Recognition, 26, 215 (2013). [ Links ]
16. K.M. Elokely, R.J. Doerksen, Docking challenge: Protein sampling and molecular docking performance, Journal of Chemical and Information Modeling, 53, 1934 (2013). [ Links ]
17. X.-Y. Meng, H.-X. Zhang, M. Mezei, M. Cui, Molecular Docking: A powerful approach for structure-based drug discovery, Current Computer-Aided Drug Design, 7, 146 (2001). [ Links ]
18. S.-Y. Huang, X. Zou, Advances and challenges in protein-ligand docking, International Journal of Molecular Sciences, 11, 3016 (2010). [ Links ]
19. O. Trott, A.J. Olson, AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, Journal of Computational Chemistry, 31, 455 (2010). [ Links ]
20. K. Morteza-Semnania, G. Amin, M.R. Shidfar, H. Hadizadeh, A. Shafiee, Antifungal activity of the methanolic extract and alkaloids of Glaucium oxylobum, Fitoterapia, 74, 493 (2003). [ Links ]
21. C.D. Hufford, Y. Jia, E.M. Croom Jr., I. Muhammed, A.L. Okunade, A.M. Clark, Antimicrobial compounds from Petalostemum purpureum, Journal of Natural Products, 56, 1878 (1993). [ Links ]
22. Y-H. Wang, A-J. Hou, G-F. Zhu, D-F. Chen, H-D. Sun, Cytotoxic and antifungal isoprenylated xanthones and flavonoids from Cudrania fruticosa, Planta Medica, 71, 273 (2005). [ Links ]
23. G. Larcher, C. Morel, G. Tronchin, A. Landreau, D. Séraphin, P. Richomme, J-P. Bouchara, Investigation of the antifungal activity of caledonixanthone E and other xanthones against Aspergillus fumigatus, Planta Medica, 70, 569 (2004). [ Links ]
24. T. Fukai, Y. Oku, A-J. Hou, M. Yonekawa, S. Terada, Antimicrobial activity of hydrophobic xanthones from Cudrania cochinchinensis against Bacillus subtilis and methicillin-resistant Staphylococcus aureus, Chemistry & Biodiversity, 1, 138 (2004). [ Links ]
25. G. Gopalakrishnan, B. Banumathi, G. Suresh, Evaluation of the antifungal activity of natural xanthones from Garcinia mangostana and their synthetic derivatives, Journal of Natural Products, 60, 519 (1997). [ Links ]
26. E. Pinto, C. Alfonso, S. Duarte, L. Vale-Silva, E. Costa, E. Sousa, M. Pinto, Antifungal activity of xanthones: Evaluation of their effect on ergosterol biosynthesis by high-performance liquid chromatography, Chemical Biology & Drug Design, 77, 212 (2011). [ Links ]
