











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La malaria es uno de los problemas de salud más graves en muchas partes del mundo, particularmente en África y América Latina porque son las regiones con más altos índices de mortalidad [1]. Esta enfermedad es causada por un parásito de género Plasmodium, transmitido a los seres humanos por la picadura del mosquito hembra del género Anopheles, que se reproduce en regiones que combinan calor, humedad y vegetación [2]. Datos recientes, estiman que la malaria está presente en 97 países y se calcula que 3200 millones de personas están en riesgo de contraer la enfermedad [3]. En el 2015 se reportaron 214 millones de casos de malaria, ocasionando la muerte de 438.000 personas, en su mayoría niños menores de 5 años; África reportó la mayoría de los casos [4]. Una de las alternativas para el control de la morbimortalidad por malaria es la quimioterapia, sin embargo, se están presentando cepas de Plasmodium resistentes a los principales tratamientos, falla terapéutica, además de un escaso acceso a los medicamentos, entre otros, lo anterior aumenta el riesgo, y a la vez complica su prevención y tratamiento [5].
Actualmente, hay descubrimientos de nuevos blancos moleculares para el diagnóstico, prevención y tratamiento de enfermedades como la malaria, también mediante el secuenciamiento del genoma y el proteoma de Plasmodium, lo que ha permitido postular nuevos modelos para el diseño de fármacos, además del desarrollo de la química combinatoria y el uso de la síntesis total de pequeñas moléculas; las cuales deben pasar por un proceso inicial de evaluación in vitro mediante técnicas que determinan la concentración a la cual se inhibe en un 50% el crecimiento del parásito (IC50) [6-7]. Una generación de moléculas activas contra Plasmodium falciparum basadas en los mecanismos de acción de los medicamentos de uso actual como la cloroquina o en procesos metabólicos que ocurren en el patógeno están siendo sintetizadas, sin embargo, aunque se han descubierto y diseñado muchas moléculas con un potencial capaz de inhibir el crecimiento del parásito en cultivo in vitro y en modelos murinos, algunos son descartados por efectos tóxicos, resistencia por parte del patógeno, poca solubilidad y biodisponibilidad [8-11], por lo cual, aún se debe continuar en la búsqueda de nuevos antimaláricos efectivos y de bajo costo que permitan su uso combinado con otros medicamentos y así evitar el desarrollo de la resistencia del patógeno a los diferentes fármacos [12].
En la presente revisión se muestran las estrategias de diseño y desarrollo de nuevos fármacos obtenidos de plantas y de origen sintético basados en los blancos terapéuticos más estudiados, las cuales fueron consultadas en varias bases de datos (Science Direct, PUBMED, SciELO consultadas en agosto de 2017), libros especializados y demás reportes bibliográficos relacionados con estudios de actividad antiplasmodial y antimalárica de compuestos activos sobre Plasmodium, lo que ha llevado a la identificación de una serie de posibles nuevos compuestos antimaláricos.
Moléculas basadas en el núcleo quinolínico
La quimioterapia ha sido posible gracias al descubrimiento de compuestos a partir de las plantas. Un alcaloide que ha jugado un papel importante y esencial en la medicina por cientos de años es la quinina, su química ha fascinado a muchos investigadores y ha motivado cambios en la química orgánica, síntesis enantioselectiva y en la química industrial moderna. La quinina se convirtió en un compuesto líder para el tratamiento de la malaria y ha sido empleado como modelo para realizar nuevos agentes antimaláricos por vía sintética con el núcleo quinolínico, como lo son: cloroquina (CQ), amodiaquina (AQ), mefloquina (MQ) y primaquina (PQ) [13].
Una nueva clase de análogos de la cloroquina con cadena lateral modificada están siendo sintetizados, Krogstad et al. en 1998 [14] sintetizaron una molécula denominada AQ-13 similar a la CQ con la diferencia del grupo metil en el carbono C-10. Esta molécula presentó una potente actividad con valores de concentración inhibitoria (IC50) entre 5-15 nM, sobre el cultivo in vitro de P. falciparum y un mecanismo de inhibición de la polimerización del grupo hemo, sin embargo, se halló resistencia similar a la CQ, estudios clínicos fase I exhiben farmacocinéticas y toxicidades levemente diferentes a la CQ [15]. La amodiaquina es un fármaco efectivo, pero, en dosis superiores presenta efectos hepatotóxicos causando agrunulocitosis, por lo que actualmente se están presentando desarrollos en el diseño y síntesis de moléculas basadas en las 4-aminoquinolinas [16]. Un análogo similar a la amodiaquina fue diseñado bajo la comprensión del mecanismo de toxicidad de este fármaco. O'Neill et al. [17] realizaron estudios regioisoméricos de la amodiaquina e intercambiaron las posiciones de los grupos hidroxi y el grupo amino, generando una nueva molécula llamada isoquina y otra N-t-butil-isoquina que son bastante activas y no generan hepatotóxicidad. Basados en las estructuras de la AQ y PQ se han derivado análogos estructurales conservando el grupo halogenuro en la posición C-7 del anillo, la cadena alquílica y la amina terciaria, todos estos derivados han presentado inhibición sobre el patógeno con similar modo de acción a la AQ y PQ.
La tafenoquina es otro compuesto que se encuentra en etapas avanzadas de desarrollo clínico, presenta un índice terapéutico más extenso que la primaquina y una eliminación mucho más lenta, pero su función terapéutica aún no se ha establecido [18]. Al mismo tiempo, el desarrollo de 2-terc-butilprimaquina ha proporcionado la primera 8-ami-noquinolina completamente libre de toxicidad de metahemoglobina y actualmente se encuentra en desarrollo preclínico. De todos los análogos de la primaquina, las 4-alquil-5-alcoxi, 4-metil-5-fenoxi y 2-terc-butil-primaquina presentan una actividad curativa, toxicidad reducida y con una mayor promesa terapéutica (ver figura 1) [19].
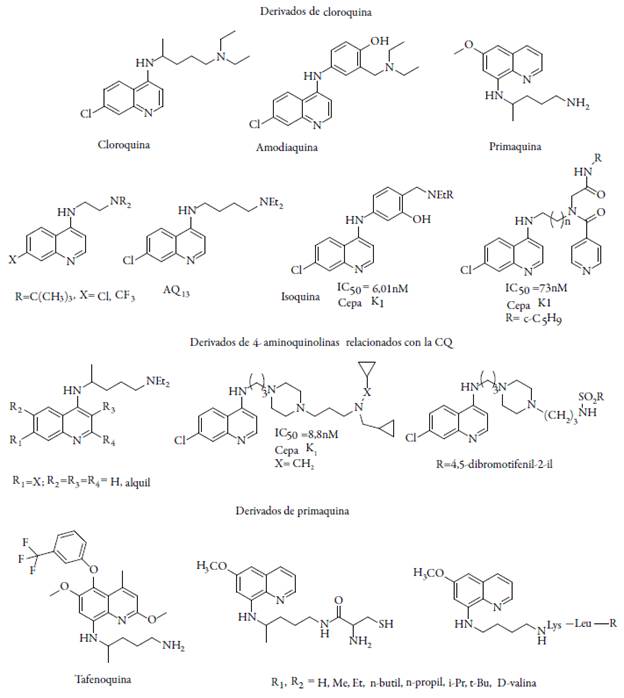
Por otro lado, es estudiada una serie de complejos de rutenio-p-areno-cloroquina [RuCl2(CQ)]2 activos frente a cepas de P. falciparum resistentes a CQ con actividad antiplasmodial; este tipo de moléculas se evalúan en cuanto a su lipofilicidad, basicidad, coeficientes de reparto, valores de pKa, habilidad de inhibir la formación del grupo hemo en interfaces agua/n-octanol con la finalidad de predecir la superior acción anti-plasmodial contra patógenos resistentes, los resultados indican tendencias interesantes como que la actividad antiplasmodial está relacionada con un equilibrio de los efectos asociados con la lipofilia, basicidad, y los detalles estructurales de los compuestos estudiados [20].
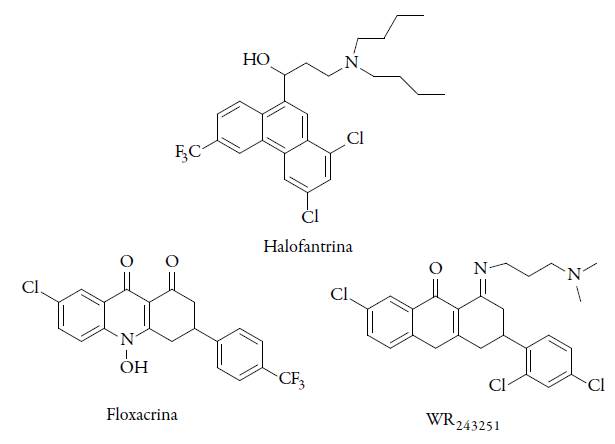
En lo que respecta a la basicidad de los antimaláricos, en diversos estudios se ha determinado que la halofantrina y sus derivados presentan un "efecto básico débil"; por lo que este tipo de principios activos tienen propiedades básicas, y se concentran en la vacuola digestiva ácida, y al tener una forma protonada no pueden atravesar nuevamente la membrana, generando toxicidad para el patógeno, pero también cardiotoxicidad en pacientes tratados. Un derivado de la halofantrina (el N-desbutilhalofantrina) es conocido como un metabolito activo de este medicamento, en el que se pudo eliminar el efecto secundario de cardiotoxicidad, sin alterar su actividad antimalárica, siendo la N-desbutilhalofantrina mucho más activa in vitro que la lumefantrina [21]. Otro ejemplo es la dihidroacridindiona WR243251 (derivado de la floxacrina), el cual fue desarrollado a partir del compuesto líder mepacrina [22], poco se conoce de este compuesto, en estudios in vivo presentó una actividad significativa contra cepas de P. falciparum resistentes a CQ en monos Aotus, además de que inhibió el desarrollo de esporozoítos de P. vivax en mosquitos; también se ha demostrado que este principio activo inhibe la biocristalización del grupo hemo, pero esto solo no representa claramente el mecanismo de su actividad, dado que se ha evidenciado que inhibe la respiración celular del patógeno. Estas moléculas son promisorias para ser candidatas antimaláricas y actualmente están en estudios de modelos in vivo. Sin embargo, el futuro potencial de esta clase de compuestos puede cuestionarse por su aparente resistencia cruzada con la CQ, lo que limito la continuidad en los estudios clínicos fase I (ver figura 2) [23].
Compuestos tipo trioxanos
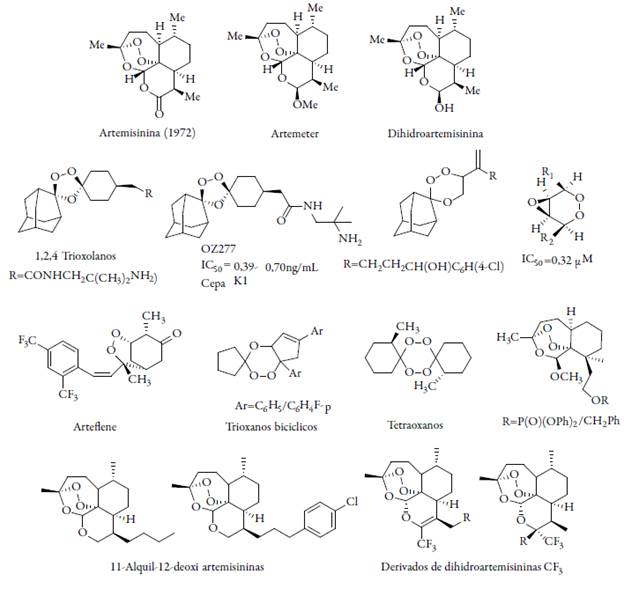
El descubrimiento y desarrollo de la artemisinina a partir de la planta Artemisia annua (Asteraceae), una hierba china conocida popularmente en este país como Qing hao y empleada por la medicina tradicional por más de 2000 años para el tratamiento de fiebres es nueva clase de antimaláricos, tiene la estructura de una sequiterpenolactona tipo trioxano con un puente endoperóxido, así se le confiere su actividad biológica, aunque es poco soluble en agua. Debido a la naturaleza altamente lipofílica y pobre solubilidad, la artemisinina presenta la desventaja de una pobre vida media en plasma, por lo que es muy probable que no se consiga eliminar el patógeno de forma completa en el organismo, de ahí la necesidad de tratamientos prolongados con este fármaco [13-24]. Por la estructura con un peróxido cíclico tipo trioxano se han sintetizado derivados basados en este modelo con rápida acción, más potentes, económicos y sin presentar casos clínicos de resistencia comparados con otros fármacos antimaláricos. Estos son compuestos prometedores, pero tienen una vida media corta por hidrolizarse rápidamente generando sustancias tóxicas [25-26]. Específicamente, el compuesto OZ277, que actualmente es la molécula líder como antimalárico con un mecanismo de acción similar al de las artemisininas, presenta ventajas como su fácil y económico proceso de síntesis, mejor biodisponibilidad y un reporte de eficacia in vivo del 99,9% de actividad a 30 mg/kg (oral) [27-28].
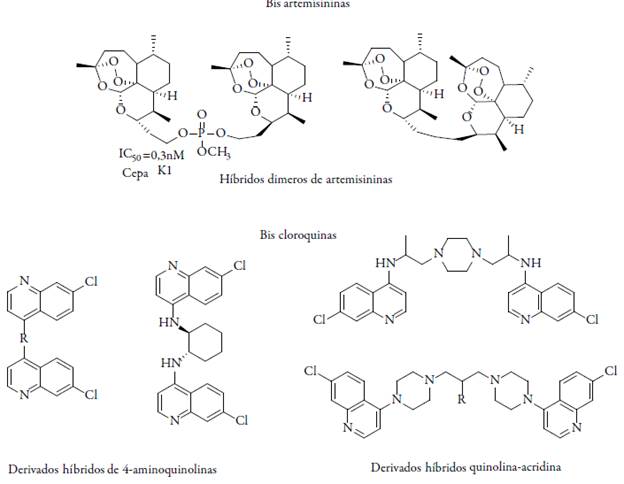
Otros estudios han tratado, aprovechando las ventajas farmacológicas, de combinar a partir de las denominadas trioxaquinas de los peróxidos con las 4-aminoquinolinas; los residuos 4-aminoquinolínicos deberían facilitar el transporte a la vacuola alimenticia, donde el FeII-Hemo liberado durante la digestión de la hemoglobina activa la unidad trioxano. La trioxaquinolina más activa sintetizada hasta el momento es la DU-1102, una molécula altamente eficiente contra cepas de P. falciparum resistentes a la CQ, posiblemente, como consecuencia de su modo de acción inespecífico; además algunos de los peróxidos mencionados exhiben actividades farmacológicas adicionales [29]. Se han sintetizado también dímeros de artemisinina como unidad central. El compuesto dimérico (ver figura 2) exhibe un valor de concentración efectiva de CE50 = 1,3 nM contra P. falciparum in vitro, mientras que artemisinina presenta un CE50 = 9,7 nM, en las mismas condiciones [30]. La simplificación del núcleo de artemisinina lleva a 3-ariltrioxanos sintéticos.
En una aplicación oral a un modelo murino, el compuesto fue dos veces más activo que la artemisinina en 48 horas. Basándose en la hipótesis de que el mecanismo de acción de las artemisininas depende de la generación de radicales libres por ruptura del puente endoperóxido, puede asumirse que otros peróxidos no relacionados estructuralmente con artemisinina, también pueden exhibir actividad antiplasmodial [31]. Otro tipo de modelos diméricos se basa en una serie de híbridos de artemisininas-vinil sulfonas con actividad potencial y capacidad de inhibición de la falcipaina 2, actúan sobre la vacuola digestiva mediante una activación del puente endoperóxido, estas son sintetizadas y evaluadas sobre cepas resistentes W2 de Plasmodium falciparum en un rango de concentración nanomolar (ver figura 3) [32].
Moléculas catiónicas y quelantes de hierro
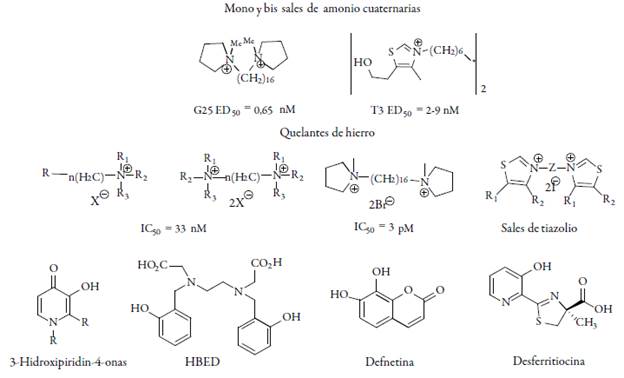
Otro tipo de compuestos activos son las moléculas catiónicas, con el nombre de G25 y T3; sales de 2-aminopiridinium, amidinas y guanidinas, han obtenido inhibiciones de crecimiento en 50% de parásitos con dosis muy inferiores (IC50=0,65 nM) a las empleadas normalmente con los medicamentos ya mencionados; estos actúan en el metabolismo de fosfolípidos inhibiendo la producción de colina que es indispensable en la síntesis y el desarrollo del patógeno, sin embargo, presentan una muy baja biodisponibilidad, dado que se degradan fácilmente en los fluidos corporales [33-34].
Estas moléculas se han ensayado en monos infectados con malaria los cuales se curan y son menos tóxicos; el G25 por otro lado, también ha mostrado su efectividad en ratones infectados con Plasmodium berghei que habían desarrollado resistencia a los antimaláricos, tanto en pruebas de laboratorio como en animales. Es por esto que nuevas moléculas amido bicíclicas y ésteres de ácidos dialquilamino fueron sintetizados y apenas están siendo investigadas como antimaláricos, actualmente, se hacen estudios de relaciones estructura-actividad donde se discutieron con más de 200 derivados [35]. En particular, los compuestos de tipo éster mostraron buena actividad antiplasmodial, uno de ellos se puso a prueba in vivo contra P. berghei con resultados prometedores, aunque el objetivo y el mecanismo de acción de estos compuestos aún se desconoce. Además, se ha logrado una clara mejora en sus propiedades antiplasmodiales [36].
Varios 4-aminobiciclos [2.2.2] octilftalato ω-ésteres de ácidos dialquilamino se han diseñado, y tienen actividad contra la polifarmacorresistente cepa K1 del P. falciparum y Trypanosoma brucei rhodesiense (STIB 900). La actividad biológica fue influenciada por la relación de la configuración en la posición 2 del anillo, por la longitud de la cadena y la sustitución de aminoácidos. Los esteres fueron tan activos como la cloro-quina, sin embargo, uno de ellos exhibe la más alta actividad y selectividad antiplasmodial de todos los derivados del biciclo-octano preparados y hasta ahora reportados (ver figura 4) [37].
Inhibidores de proteasas y moléculas peptídicas
Otras moléculas exploradas con actividad antiprotozoaria son DB-75 y DB-289 (2,5-bis(4-amidinofenil) furanos) las cuales son agentes tripanocidas, químicamente son diamidinas aromáticas que originalmente se desarrollaron como estructuras análogas de la pentamidina que es antitripanosomal, estos compuestos presentaron actividad contra P. falciparum. Otro caso es el compuesto PB-93 empleado como agente antitumoral, este inhibe la proteína farnesiltransferasa y el cual se ha determinado que es clave para la supervivencia de P. falciparum [38-41]. La farnesiltransferasa es una proteína heterodimérica que cataliza la transferencia de un residuo farnesil desde farnesilpirofosfato hacia una cisteína de una cadena lateral cercana al extremo carboxiterminal de algunas proteínas. En los últimos años se desarrollaron inhibidores de farnesiltransferasa como potenciales fármacos en terapias anticáncer y como posibles antimaláricos (ver figura 4) [42-43]. Por otra parte, la síntesis y evaluación biológica de una nueva clase de inhibidores de Falcipain-2 (FP-2) peptidomiméticos I que son una familia papaína-(C1A) cisteína proteasa, juegan un papel importante en el ciclo de vida del patógeno por ser degradantes de las proteínas del eritrocito y que contienen fracciones de esteres vinílicos con alto potencial (R1 = Ven, CONMe2, CO2Me para R = CH2CH2Ph, H; R1 = NC, R = H).
El perfil de reactividad de vinilo éster I (R = H, R1 = CO2Me) demostró ser muy potente y selectivo inhibidor de falcipain-2 [44-45]. Otros compuestos como los ciclodepsipéptidos mostraron una interesante gama de actividad biológica. Los miembros de esta nueva clase de potenciales fármacos tienen una característica común de ciclodepsipéptidos, además, se basan en las interacciones con distintos compartimentos celulares y vías de transducción de señales. Algunos ciclodepsipéptidos están siendo evaluados en estudios clínicos y, actualmente, se utilizan en la terapia del cáncer, generalmente en combinación con otros fármacos citotóxicos [46].
Una serie de derivados de la neocriptoleptina como los cloro-amino-alquilamino-sus-tituidos (5-metil-5H-indolo [2,3-b] quinolina) fueron sintetizados y evaluados como agentes antiplasmodiales. Esta evaluación también incluyó la citotoxicidad en células MRC5; la inhibición de la formación de la β-hematina, y las interacciones de ADN y la introducción de cadenas amino-alquilamino aumento sustancialmente la actividad antiplasmodial de la neocriptolepina. Las formas más eficientes de estos compuestos mostraron actividades antiplasmodiales en el rango nanomolar. La degradación de la hemoglobina en la vacuola alimenticia del patógeno se cataliza en forma dirigida por una serie de proteasas, las cuales están parcialmente caracterizadas y disponibles como proteínas recombinantes. La ruptura inicial de la hemoglobina está mediada por las proteasas aspárticas plasmepsina I, II y IV. Nuevos inhibidores de proteasas están siendo diseñados, como el N1, N1-dietil-N4-(5-metil-5H-indolo [2,3-b] Hidroxi-8-il) pentano-1 ,4-diamina (I) el cual mostró una IC50 de 0,01 mM y un índice de selectividad de 1800 (ver figura 5) [47].
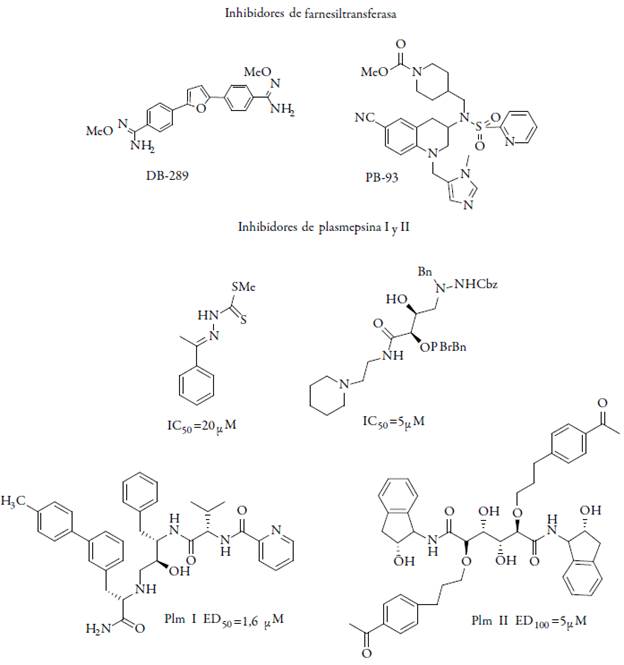
Inhibidores de la síntesis de ácidos grasos
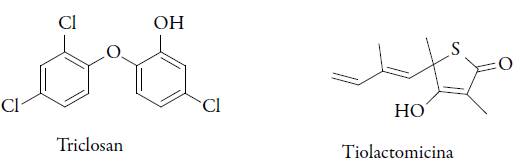
En los parásitos de Plasmodium ha sido demostrada la existencia de un sistema de bio-síntesis de los ácidos grasos tipo II. En este caso, las enzimas individuales están localizadas en el interior del apicoplasto y son similares a las involucradas en la síntesis de isoprenoides. El antibiótico natural tiolactomicina inhibe las enzimas Fab B, Fab F y Fab H, las cuales catalizan las distintas etapas de condensación en la síntesis de ácidos grasos en P. falciparum. El compuesto es activo in vivo contra el crecimiento de P. falciparum (CE50 = 50 μM). A su vez, el triclosan es significativamente más activo que la tiolactomicina frente a P. falciparum; este es un inhibidor de la trans-2-enoil-ACP-reductasa, Fab I (CE50 = 1 μM). Además, se demostró su eficacia < 90% en ratones infectados con P. berghei. Los correspondientes valores de CE50 y LD90, tras inyecciones subcutáneas, fueron 3 y 30 mg/kg respectivamente (ver figura 6). Se testaron varios derivados de triclosán en cuanto a su actividad antimalárica, sin embargo, como presenta actividad frente a un amplio espectro de bacterias, se utiliza como conservante en varios productos de uso doméstico, pero no se aconseja su uso para el tratamiento por vía oral [48].
Inhibidores de la glicólisis
El ciclo funcional del ácido cítrico está ausente en los parásitos de la malaria, por lo tanto, su energía metabólica depende mayoritariamente de la glicólisis anaerobia. El NAD+ debe ser regenerado por reducción de piruvato a lactato. La enzima lactato deshidrogenasa de P. falciparum ha sido producida como una proteína recombinante, y se ha hecho un análisis cristalográfico de alta resolución de ella. Se está utilizando una combinación de métodos de evaluación biológica de alto rendimiento y diferentes técnicas de diseño racional con el fin de identificar inhibidores específicos que no afecten esta enzima en humanos (ver figura 7) [49-50].
Sin embargo, a pesar de ser un blanco específico en el patógeno y que todas estas nuevas moléculas han presentado una capacidad de inhibir el crecimiento del parásito en cultivo in vitro, varias de ellas son descartadas por sus efectos tóxicos, resistencia del parásito, pobre solubilidad y biodisponibilidad, por lo que aún se debe continuar la búsqueda de nuevos antimaláricos que permitan su uso combinado con otros medicamentos para así evitar el desarrollo de la resistencia del patógeno [51].
Moléculas híbridas
Una tendencia que está tomando fuerza en la actualidad en el diseño de nuevos agentes antimaláricos son las moléculas híbridas, las cuales están basadas estructuralmente en la unión de farmacóforos con procedimientos sintéticos de bajo costo y bajo potencial para el desarrollo de la resistencia [52]. Actualmente las moléculas híbridas son consideradas como un nicho en el diseño racional de fármacos; dichas moléculas se definen como entidades químicas con dos (o más) dominios estructurales que presentan diferentes funciones biológicas (estructura quimérica también es una denominación, pero se prefiere el termino de híbridos), en la cual la doble actividad indica que una molécula híbrida actúa como dos farmacóforos distintos [53]. Ambas entidades de la molécula híbrida no necesariamente actúan sobre el mismo blanco biológico. La estrategia de una molécula híbrida mejora la actividad biológica en un fragmento de la molécula, lo cual se da mediante la adición de funciones químicas capaces de interactuar con las regiones adyacentes de la molécula activa cuando la proteína destino se desconoce. El fragmento de auto-ensamblaje consiste en permitir que dos o tres fragmentos con funcionalidades diferentes o semejantes puedan reaccionar en el sitio de destino para generar in situ enlaces covalentes con distintos blancos.
En esta dirección se ha diseñado mediante metodologías de síntesis una nueva molécula híbrida entre dos potentes antimaláricos, la artemisinina y la quinina (artemisnina-quinina). Resultados con esta molécula híbrida mostraron una mejor actividad (IC50=8,95 nM) en comparación con las moléculas individuales de artemisinina (IC50=49,4 nM), quinina (IC50=149 nM) e incluso con la combinación 1:1 de artemisinina y quinina (IC50= 31,8 nM) cuando fueron evaluadas en cepas 3D7 de P. falciparum sensibles a CQ[54]. Por otra parte, Cowman et al. han diseñado moléculas híbridas denominadas bisquinolinas, en las cuales dos núcleos quinolínicos están conectados por varios enlaces, siendo mucho más activas y con mecanismos de acción similar a la CQ, pero fueron tóxicas en estudios in vivo, por lo que actualmente son el motivo de varios trabajos de investigación [55-56]. También se han sintetizado modelos híbridos de trioxaquinas con un trioxano como estructura base (que es responsable de la actividad antimalárica de la artemisinina), ambos unidos a una entidad aminoquinolina (que es responsable de la propiedad antiplasmodial de la cloroquina). Estas trioxaquinas híbridas son muy potentes y exhiben actividad eficaz contra los fases eritrocíticas de cepas de P. falci-parum resistentes a CQ [57]. Musonda et al. [58-60] han diseñado híbridos a partir de dos medicamentos conocidos contra la malaria, la cloroquina (CQ) y el astemizol, encontrándose que poseen mejor actividad in vitro e in vivo contra cepas de Plasmodium resistentes a CQ.
Sunduru et al. [61] han funcionalizado las 4-aminoquinolinas con oxalamidas y triazinas en la cadena lateral, y fueron evaluados y seleccionados por sus actividades contra la malaria. En un ensayo in vitro, uno de los derivados de la triazina resultó ser el más activo contra la cepa 3D7 de Plasmodium falciparum sensible a CQ, presentándose un valor de IC50 de 5,23 ng/mL; mientras que, un derivado de la oxalamida presento in vivo supresión de la parasitemia del 70,45 % en el día 4 contra la cepa N-67 de Plasmodium yoelii resistente a CQ, demostrando que la estructura-actividad está relacionada con una función amida secundaria que aumenta la actividad, mientras que la presencia de una amida terciaria y un α-cetoamida disminuye la actividad debido a que también disminuye la capacidad de los sitios de unión con el sitio activo. Por otro lado, derivados de triazina, sustituidos con aminas que tienen nitrógeno terminal de base como la clo-roquina, aumentan potencialmente la actividad antiplasmodial.
Recientemente, se ha desarrollado una innovadora clase de nuevos híbridos basados en productos naturales. Hans et al. [62] sintetizaron 36 nuevas moléculas híbridas a partir de productos naturales basados en β-amino alcoholes, tiolactona-chalconas y isatin-chalconas; donde las moléculas híbridas tiolactona-chalconas mostraron valores de IC50 en un rango de 0,68 a 6,08 (M, encontrándose la mayor actividad frente a la cepa W2 de P. falciparum; mientras que con las de isatin-chalconas se encontró una IC50 de 14,9 μΜ o menos. En otro estudio se sintetizaron 34 derivados de lupeol, demostrando actividad contra la malaria en una dosis más baja (10 μg/mL) que la del lupeol puro (25 μg/mL) [51]. Este número limitado de ejemplos con moléculas híbridas citadas está tomando fuerza en la investigación, por lo que son muchas otras nuevas entidades que se han reportado, por ejemplo, híbridos de artemisinina-acridina, trioxaferroquinas, cloroquina-astemizole [63] triazinas 9-anilinoacridina, arteminin-dipetidil vinil sulfonas [64], aminoquinolinas híbridas [65] entre otras; en su mayoría, estas mejoran el efecto antiplasmodial e ilustran el concepto de moléculas híbridas y sus potenciales aplicaciones (ver figura 8).
Derivados indólicos
La criptolepina componente presente en gran cantidad en la especie Cryptolepis sanguinolenta, y en un estudio presentó un IC50 de 0,134 μM sobre cepa K1 de P. falciparum, esto es dos veces más potente que la cloroquina (IC50 de 0,230 μM), sin embargo, presento un alto grado de citotoxicidad por su capacidad de intercalarse en el ADN. Otro compuesto en este grupo es el 3-metilen-sustituido (ver figura 9), el cual es usado en la evaluación de indolinonas como inhibidores de la falcipaína, una enzima usada por el parásito para digestión de hemoglobina. Estos compuestos reaccionan fácilmente con tioles a través de un mecanismo de adición-eliminación, lo que indica su potencial como inhibidores de enzimas como la cisteín-proteasa (falcipaina). Varias indolinonas que contienen un fragmento de reconocimiento Leu-i-amilo son inhibidoras moderadas del P. falciparum y de la proteína cisteín-proteasa falcipaína-2, pero no de la proteasa relacionada falcipaína-3, estos compuestos muestran la actividad antiplasmodial contra la cepa W2 de P. falciparum resistente a la cloroquina en el rango micromolar [66].
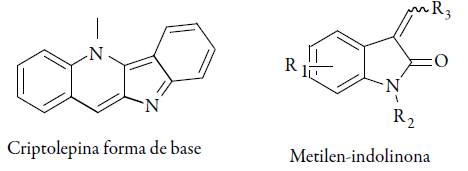
Derivados de productos naturales
Existe gran variedad de metabolitos secundarios que son químicamente diversos, los cuales pueden ser obtenidos a partir de plantas específicas reportadas por la medicina tradicional, o a nivel de género o familia botánica, y muchos compuestos de este tipo han sido validados como antimaláricos promisorios, puesto que en bioensayos hay actividad antiplasmodial significativa frente a diferentes cepas del patógeno; algunos compuestos se muestran en la figura 10[67]. El yingzhaosu A, un peróxido aislado de la planta Artabotrys uncinatus, L. (Annonaceae), y su derivado sintético artefleno, también son activos, sin embargo, su pobre eficacia clínica y la dificultad de su síntesis, hacen difícil la continuación de su desarrollo [68].
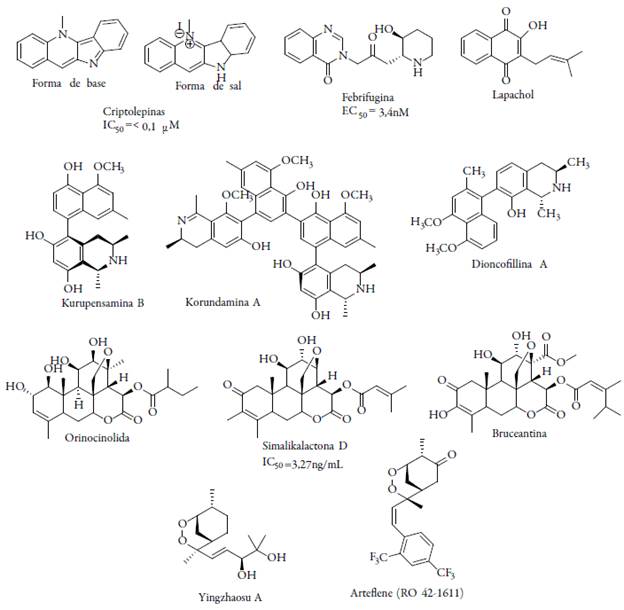
Entre los productos naturales con potencial actividad antiplasmodial, los alcaloides indólicos representan un grupo de compuestos de interesantes farmacológico; los indoles son compuestos orgánicos heterocíclicos, con estructura bicíclica que consisten en un anillo de seis miembros (benceno) unido a otro de cinco miembros (pirrol), donde participa un par aislado de electrones de nitrógeno en anillo aromático, por lo que refiere a que el indol no es una base y no representa una amina simple. Uno de los alcaloides más conocidos es la quinina, cuyo núcleo quinolínico se forma por la vía biosintética común de alcaloides indolomonoterpénicos (la clase más importante de indol alcaloides).
Uno de los alcaloides que ha mostrado interés en su descubrimiento es el alcaloide indoloquinolínico criptolepina, el cual es el componente con mayor proporción aislado de las raíces y hojas de Cryptolepis sanguinolenta, una planta comúnmente usada por la medicina tradicional en el oeste de África para el tratamiento de la malaria y otras enfermedades. Esta molécula mostró ser activa en cepas K1 y T996 de P. falciparum resistentes y sensibles a la cloroquina. Este alcaloide con características débilmente básicas es activo, mientras que otros alcaloides estructuralmente relacionados con perfiles de ácido-base son inactivos. En modelo in vivo en ratones infectados con P. bergheiyoelii mostraron una significativa reducción de la parasitemia cuando se administra oralmente a dosis de 50 mg/kg de peso por cuatro días. Sin embargo, como resultado de su capacidad de intercalarse en el ADN, la inhibición de replicación y trascripción del ADN, la criptolepina es tóxica [69].
La síntesis de la criptolepina es bastante viable y gran variedad de análogos sintéticos han sido reportados; entre los diferentes derivados sintéticos obtenidos a partir de la criptolepina, la 2,7-dibromocriptolepina es la más activa, presentando una actividad aproximadamente 10 veces mayor que la criptolepina con una inhibición del 89 % en una sola dosis de 12 mg/kg/día por vía intraperitoneal y una citotoxicidad apenas un poco mayor contra células cancerígenas, y baja toxicidad en ratones (ver figura 10) [70].
CONCLUSIONES
A pesar de los grandes esfuerzos por controlar la enfermedad de la malaria, aún no existen vacunas o medicamentos capaces de prevenirla, solo existen tratamientos curativos que presentan inconvenientes como: la eficacia, la toxicidad variable y la resistencia del parásito a ellos, lo que hace que el control de la enfermedad se convierta en uno de los principales desafíos para la salud pública. En las últimas dos décadas, nuevos candidatos antimaláricos han sido desarrollados, pero en el descubrimiento de nuevos fármacos para contribuir al tratamiento de distintas enfermedades (entre estas la malaria), se debe tener en cuenta que estos sean: potentes, selectivos, seguros, no presenten resistencia, tolerables, no tóxicos, aplicables por diferentes vías, estables en líquidos biológicos y que se puedan obtener a gran escala industrial. El proceso de investigación y desarrollo de un fármaco es largo y complejo, involucra grandes costos y pocas posibilidades de éxito, debido principalmente a los efectos tóxicos y a los parámetros farmacocinéticos los cuales están sujetos a una gran variabilidad interindividual.
En los últimos años se han realizado considerables esfuerzos en diseñar moléculas revirtiendo la resistencia a los medicamentos, nuevas clases estructurales de agentes antimaláricos con núcleo quinolínico, trioxanos, quelantes de hierro, sales de amonio cuaternario, inhibidores de proteasas, glicólisis y ácidos grasos, nuevos híbridos, péptidos, indoles y derivados de productos naturales.
La investigación realizada sobre las quinolinas y las artemisininas proporcionó información importante con respecto a la relación estructura-actividad de esta clase de compuestos, además, existe una tendencia creciente hacia el desarrollo de antimaláricos con blancos específicos, por ejemplo, agentes inhibidores del grupo hemo, de la farne-siltransferasa, inhibidores de lactato deshidrogenasa, o la formación de híbridos antimaláricos. De las muchas moléculas identificadas y ensayadas aún no se presenta una nueva generación de candidatos antimaláricos dado que están siendo descartados en las distintas etapas del proceso, por lo que es urgente continuar la búsqueda de nuevos antimaláricos efectivos y de bajo costo que permitan su uso combinado con otros medicamentos y así evitar el desarrollo de la resistencia del parásito a los diferentes fármacos.
La complejidad de estos procesos es manejada por una diversidad de disciplinas científicas que incluye químicos orgánicos, biólogos moleculares, toxicólogos, médicos, farmacólogos, bioquímicos y científicos de la computación que participan en alguna etapa del proceso, lo que en parte explica los enormes costos involucrados. Finalmente, es necesaria la investigación en el desarrollo de nuevos antimaláricos para los próximos años porque la carga de la malaria seguirá siendo alta en las próximas décadas junto con la pérdida repetida de antimaláricos para la resistencia.

