











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La resistencia bacteriana se ha convertido en uno de los grandes temores para la terapia antibiótica, fármacos que eran eficaces han caído en desuso debido a la rápida capacidad de mutación que tiene el sistema de resistencia bacteriana. Aunque los antibióticos beta-lactámicos aún siguen siendo considerados de primera elección en el tratamiento de algunos procesos infecciosos [1], es apremiante la búsqueda de nuevas alternativas que ofrezcan una mayor eficacia frente a estos microorganismos. Apoyando lo anterior, hemos considerado de interés realizar estudios computacionales que permitan llegar a propuestas sólidas para el diseño posterior de moléculas que puedan resultar de interés en el área de los antiinfecciosos.
En el grupo de los antibióticos betalactámicos, se encuentra la familia de las cefalosporinas [2], las que inhiben la síntesis de la pared celular bacteriana a través de la inhibición competitiva de la enzima transpeptidasa, también llamada proteína de unión a penicilina (PBP), imposibilitando su mecanismo de acción catalítico en la unión y entrecruzamiento de las cadenas de peptidoglucano, glucoproteína que le confiere la forma, rigidez y estabilidad a la membrana celular bacteriana [3]. Sin embargo, las bacterias en su mecanismo de resistencia involucran la ruptura del anillo betalactámico por la acción de las betalactamasas [4]. Ejemplo de lo anterior es la E. coli resistente a antibióticos tipo cefalosporinas por la producción de enzimas betalactamasa de espectro extendido (BLEE), principal mecanismo de resistencia antibacteriana [5].
Las infecciones bacterianas por agentes etiológicos multiresistentes limitan el uso de los antibióticos betalactámicos, inconveniente que se acentúa con la reducción en el descubrimiento y desarrollo de nuevas moléculas con actividad antibiótica [6]. Por ello es necesario implementar otras alternativas terapéuticas que pueden ser poco eficaces conduciendo a aumentos en las tasas de morbimortalidad, así como en los costos hospitalarios [5]. Una posibilidad que puede contribuir a disminuir la magnitud del problema radica en el diseño de nuevos fármacos análogos estructurales de las cefalosporinas, que surjan de un estudio computacional que permita proponer moléculas que presenten alta afinidad para inhibir la enzima PBP y que disminuyan su afinidad por la enzima BLEE [7].
El objetivo de este estudio es proponer nuevos compuestos derivados de cefalosporinas con potencial actividad antibacteriana frente a cepas de Escherichia Coli, utilizando como estructura base cefotaxima (ver figura 1), con el propósito de seleccionar moléculas que presenten mayor afinidad como inhibidores de las enzimas PBP y a su vez disminuyan o no tengan afinidad por las enzimas BLEE; para la búsqueda de moléculas con dichas características, se realizó un estudio teórico de moléculas análogas de cefalosporinas con modificaciones estructurales en la cadena lateral 7-acilamino, como se ilustra en la tabla 1, la información se obtuvo de descriptores electrónicos que permitieron comprender la relación entre las estructuras de las moléculas propuestas, su reactividad y predecir su comportamiento en el mecanismo de acción antibacteriana. También se calcularon los valores de energía libre de unión por las proteínas PBP y BLEE y se identificaron las interacciones existentes entre cada uno de las moléculas propuestas y en el sitio activo de estas enzimas, diseñándose de esta manera compuestos derivados de cefalosporinas con potencial actividad antibacteriana más efectiva y posibles candidatos teóricos para estudios de desarrollo de nuevos fármacos.
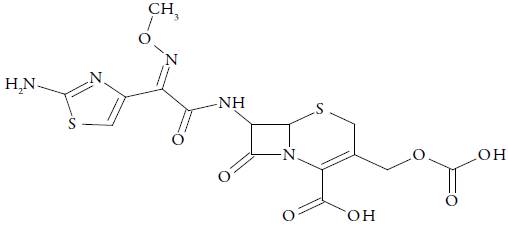
METODOLOGÍA
La metodología de este trabajo se desarrolló en dos subsecciones: en una se describe como se calcularon los descriptores de reactividad a compuestos análogos de cefalosporina; en la otra sección se describe el estudio de las interacciones entre las moléculas propuestas y el sitio activo de PBP y BLEE de E. coli realizando cálculos de acoplamiento molecular.
Estudio teórico para calcular descriptores de reactividad a los 20 análogos de cefalosporina
Se diseñaron 20 moléculas (MP) con diferentes modificaciones estructurales, utilizando como molécula referencia la cefotaxima (MREF) (ver tabla 1), se utilizó el programa GaussView 5.0 [8], para la construcción de estas moléculas, y sus geometrías se optimizaron la teoría del funcional de la densidad (DFT) [9] a un nivel X3LYP [10] basado en el cálculo DGDZVP [11]. Una vez optimizadas las moléculas se procedió a realizar los cálculos energéticos de los orbitales moleculares HOMO y LUMO (orbitales frontera), para luego hallar la dureza y suavidad global. Todos estos cálculos se realizaron con el paquete de programas Gaussian 09 [12]. Por último se realizó el cálculo del mapa del potencial electrostático molecular (MEP) para los archivos de salida de las moléculas optimizadas empleando el programa Avogadro versión 1.0 [13].
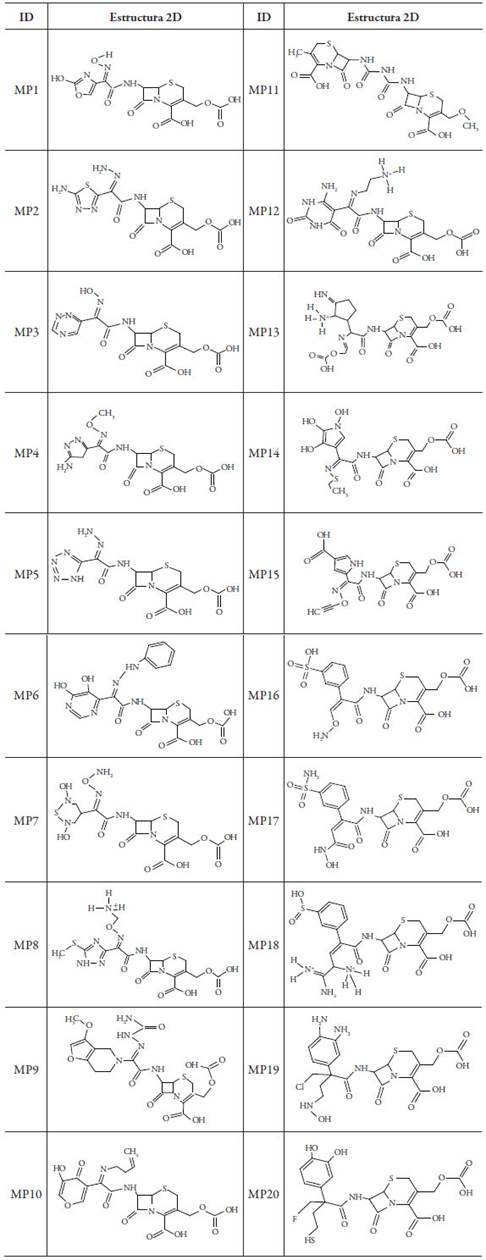
Acoplamiento molecular y estudio de interacciones
Preparación de proteínas y ligandos: la estructura 3D de la proteína PBP de una cepa de E. coli (PDB: 3MZE) en forma de complejo co-cristalizado con el ligando nativo cefoxitina (DB0133) y la estructura 3D de la BLEE (PDB: 3HLW) en forma de complejo co-cristalizado con el ligando nativo cefotaxima (DB00493), se descargaron de la base de datos PDB (Protein Data Bank) [14]. Se visualizaron y se identificaron las proteínas con sus respectivos ligandos nativo en el sitio activo con el programa SYBYL-X v2.1 [15], de allí se extrajo la estructura del ligando nativo, luego se eliminando el agua y cofactores con presentes en los complejos cristalizados, y finalmente, se adicionaron todos los hidrógenos y se asignaron las cargas Gasteiger-Marsilli [16]. Por otra parte, el ligando nativo fue preparado en AutoDocktools v1.5.6 [17], se adicionaron los hidrógenos polares, e identificaron los enlaces rotables y las cargas de Gasteiger-Marsilli.
Los parámetros del acoplamiento con AutoDock v4.2 [18] fueron: para la proteína PBP, caja del Grid centrada en el sitio de unión del ligando nativo con un tamaño 51 x 51 x 51 A, con espaciado entre los puntos de 0,375 A, generando 100 corridas por ligando; y para la proteína BLEE caja del Grid centrada en el sitio de unión del ligando nativo con un tamaño 41 x 41 x 41 A, con espaciado entre los puntos de 0,375 e igualmente estipulando 100 corridas por ligando. Los resultados de energía de unión son expresados en kcal/mol.
Los ligandos nativos previamente preparados se reacoplaron en sus respectivas proteínas, con el objetivo de validar los protocolos de acoplamiento. Se comparó la estructura cocristalizada del ligando nativo versus la estructura acoplada en el sitio activo de la proteína y se utilizaron como criterios de validación la orientación en el sitio activo de los ligandos, la energía de unión y la desviación estándar cuadrática media (RMSD). Una vez el protocolo de acoplamiento molecular descrito arriba fue validado, de tal forma que la posicion y orientacion tomada por los ligandos en la estructura 3D, fue reproducida con cierta exactitud por AutoDock 4.2, los demas compuestos análogos de la cefotaxima fueron optimizados y acoplados siguiendo el mismo protocolo. Finalmente, se seleccionó como mejor conformación aquella que presentó la menor energía libre de unión. Adicionalmente se usó el programa Discovery Studio [19] para visualizar la mejor conformación para cada ligando, identificando la ubicación del farmacóforo y el tipo de interacciones proteína-ligando en el sitio activo de la enzima.
RESULTADOS Y DISCUSIÓN
La sección de resultados y discusión se presenta de la siguiente manera: primero, se presentan los resultados de los descriptores de reactividad y el análisis de los mapas de potencial electrostático. Segundo, análisis de las interacciones resultado del acoplamiento molecular.
Descriptores de reactividad a los 20 análogos de cefalosporina
Teniendo en cuenta los efectos estéricos y electrónicos de los diferentes grupos sustituyentes sobre la reactividad del anillo betalactámico fue necesario un estudio de las propiedades electrónicas de estas moléculas. En la tabla 2 se muestran los cálculos energéticos de los orbitales fronteras HOMO y LUMO, dureza y suavidad global para las diferentes moléculas propuesta (MP) y la molécula de referencia (MREF). Se aprecia para la MREF (cefotaxima) una suavidad de 0,227 eV, que al compararse con la de las moléculas propuestas se concluye que MP2, MP6, MP9, MP11, MP13, MP14, MP19 y MP20 fueron las moléculas más suaves indicando que presentan mayor reactividad que la cefotaxima y tendencia a cambios en su población electrónica, actuando como moléculas nucleófilas o electrófilas.
Tabla 2 Descriptores globales calculados para compuestos análogos de cefalosporinas a un nivel X3LYP/DGDZVP.
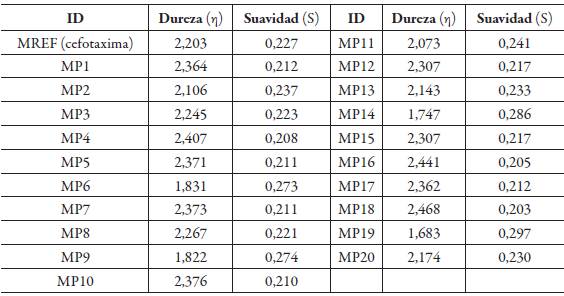
Mientras que las moléculas MP1, MP3, MP4, MP5, MP7, MP8, MP10, MP12, MP15, MP16, MP17 y MP18 presentaron una dureza mayor que el valor de referencia calculado (2,203 eV), por lo que posiblemente presentarán menor reactividad con respecto a la cefotaxima. Las moléculas pareadas MP1 y MP17, MP5 y MP7, MP12 y MP15 no presentan cambios significativos en los valores de dureza y suavidad, posiblemente debido a que el ambiente químico alrededor de R es muy similar.
Otra de las propiedades electrónicas calculadas fue el mapa de potencial electrostático (MEP), utilizado tradicionalmente en la interpretación cualitativa de la reactividad química pero que también es útil para analizar interacciones intermoleculares, debido a la identificación de regiones de potencial negativo, de alta densidad electrónica o zona nucleófila susceptible a ataque electrofílico; y de regiones o zonas de potencial positivo, de baja densidad electrónica susceptibles de ataque nucleofílico, es decir, zonas donde la molécula puede interactuar ganando electrones [20]. El potencial electrostático varía de zonas más negativas (rojo) a zonas más positivas (azul) de la siguiente manera: rojo, naranja, amarillo, verde y azul. Donde los colores intermedios indican un balance electrónico homogéneo, es decir zonas con baja densidad de carga [21].
En la figura 2 se muestran los resultados obtenidos del mapa de potencial electrostático molecular (MEP), para identificar las zonas de reactividad o zonas capaces de generar interacciones intermoleculares. Según los resultados mostrados en el mapa de potencial electrostático (MEP) de la figura 2, tanto el núcleo cefalosporánico como el sustituyente 3-acetoximetilo de dichas moléculas no presentan diferencias significativas en el potencial electrostático, con respecto a la MREF, por el contrario, los sustituyentes de la cadena lateral 7-acilamino para las MP presentan diferencias en los potenciales electrostáticos sobre estas zonas, demostrado por el cambio de colores observados en los MEP.

Figura 2 Mapa del potencial electrostático molecular (MEP) para las moléculas MREF, MP9, MP16 y MP17.
Para la molécula MP9 predominan grupos electrófilos en la cadena 7-acilamino, indicando que forman interacciones electrostáticas con grupos nucleófilos o donadores de electrones o alta formación de puentes de hidrógeno con grupos que presenten electrones libres, aun así, se observaron regiones con zonas de potencial negativo o de alta densidad electrónica. En el caso de la molécula MP16 se observa una gran zona verde, mostrando distribución electrónica homogénea o baja polaridad en los grupos sobre los que se observa ese potencial, prediciendo mayor facilidad para la formación de interacciones con residuos hidrofóbicos.
Por otro lado, la gran mayoría de las moléculas propuestas presentaron diversas zonas con potencial negativo y potencial positivo sobre la cadena 7-acilamino, es decir, zonas nucleófilas y electrófilas que crean una alta polaridad en la región, por lo que se favorecería la formación de diversas interacciones electrostáticas, ejemplo de una de ellas es la molécula MP17, donde las zonas de potencial negativo se encontraban sobre los oxígenos carboxílicos y enlazados a nitrógeno. Las zonas de potencial positivo se encontraban en cadenas alquílicas y sobre nitrógenos enlazados a átomos más electronegativos, esperando formación de interacciones con restos polares como puentes de hidrogeno. Por tanto, se infiere que estas moléculas tienen alta capacidad de formar interacciones electrostáticas con residuos de aminoácidos en el centro activo de las enzimas, favoreciendo acoplamiento ligando-receptor.
Acoplamiento molecular y estudio de interacciones
Según el alineamiento mostrado en la figura 3, siguiendo el criterio visual, se observa que la conformación resultado del docking se ubica en el sitio de unión de la PBP (PDB: 3MZE), tomando una conformación semejante a la que presenta el ligando nativo en su estructura cristalizada, igualmente ocurre en el sitio de unión de la BLEE (PDB: 3HLW). Según el criterio RMSD para ambas proteínas, se obtuvo un valor de 1,56 A para la conformación de menor energía de unión, considerándose como una conformación aceptable [22]. Por lo tanto, se puede afirmar que AutoDock v4.2 reproduce adecuadamente la forma de unión y las interacciones presentes entre ligandos nativos y proteínas.
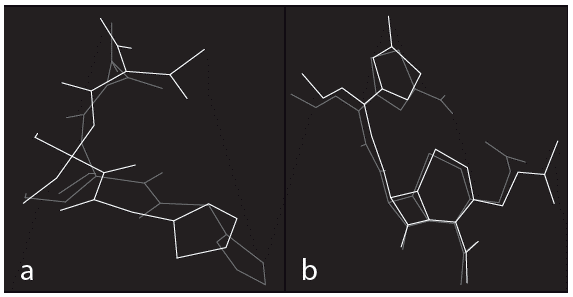
Figura 3 Alineamiento entre el ligando nativo presente en el complejo cocristalizado (en color verde) y el ligando nativo resultante del acoplamiento molecular (en color rojo), utilizando el programa AutoDock v4.2. a) con la enzima PBP b) con la enzima BLEE.
Posteriormente se llevó a cabo un segundo acoplamiento entre estas mismas moléculas y las enzimas BLEE y la enzima PBP con el fin de seleccionar aquellas que presenten baja afinidad donde su grupo farmacofórico no interacciona con los aminoácidos catalíticos del sitio activo, responsables del mecanismo de resistencia antibacteriana. Se observó que trece de las veinte moléculas presentaron menor afinidad teórica por la enzima, en comparación con el ligando nativo (cefotaxima), lo que supone menor susceptibilidad al mecanismo de resistencia y por tanto mayor eficacia antibacteriana [23], como se puede observar en la tabla 3.
Tabla 3 Afinidad (energía de unión) entre compuestos análogos de cefalosporinas y las enzimas PBP y BLEE.
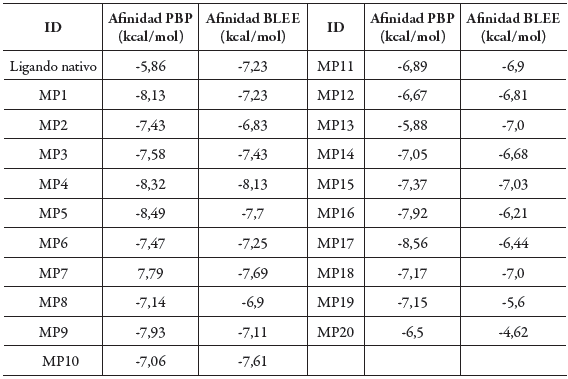
Según los valores de energía de unión se consideran a las moléculas MP9, MP16 y MP17 como las de mayor afinidad por la enzima PBP y con menor afinidad por la enzima BLEE, como moléculas de interés para el estudio de la orientación del grupo farmacofórico y las interacciones entre el ligando y el sitio activo de las proteínas PBP y BLEE. En la figura 4 se muestran las interacciones presentadas entre las moléculas MP9, MP16, MP17 con la enzima PBP. Para MP9 se identifica la cercanía entre el resto nucleófilo del residuo catalítico SER44 y el grupo farmacofórico, demostrando correcta orientación en el sitio activo y por tanto mayor probabilidad de interacción nucleofílica.
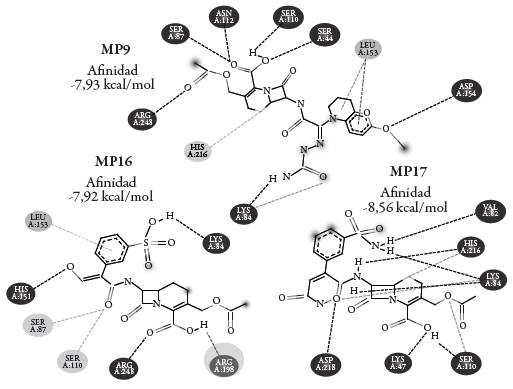
Otros aminoácidos interactuantes son SER110, SER87, ASN112, ARG248, HIS216, LYS84, ASP154 Y LEU 153, caracterizándose interacciones en su mayoría tipo puente de hidrogeno y una interacción hidrofóbica. Para la molécula MP16 se muestra una situación similar, mostrando aproximaciones de los aminoácidos catalíticos SER87 y SER110 con el grupo farmacofórico, lo que posiblemente origina posteriores interacciones y con esto el ataque nucleofílico enzimático. Otros aminoácidos interactuantes son HIS151, LEU153, LYS84 y ARG248. En cuanto a MP17 se distingue que la orientación de la conformación no favorece las interacciones deseadas con el sitio activo de la enzima, sin embargo, se observa interacción con el residuo catalítico SER110 que conllevaría a un ataque nucleofílico con el grupo de interés; otras interacciones presentadas son del tipo puente de hidrógeno: HIS216, LYS47, LYS84, ASP218 y VAL82.
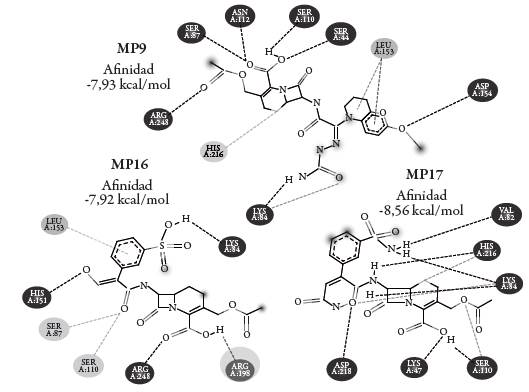
En la figura 5 se muestra las interacciones entre las moléculas MP9, MP16, MP17 con la enzima BLEE, donde se identifica que MP9 y MP16 no evidencian aproximación para interacción farmacofórica con los aminoácidos catalíticos del sitio activo (SER130, SER237 y SER274), lo que sugiere impedimento del ataque nucleofílico y con ello bloqueo de la interacción causante de la resistencia antibacteriana; estas moléculas adoptan orientaciones que favorecen la formación de interacciones de la cadena 7-acilamino con los restos del sito activo, en lugar del farmacóforo o regiones cercanas al anillo betalactámico, Por el contrario la molécula MP17 presentó interacción favorable del grupo farmacofórico con el residuo SER274, produciendo la inactivación del anillo betalactámico, hecho que predispone a la aparición de resistencia antibacteriana.
CONCLUSIONES
Los resultados de los cálculos de suavidad permiten concluir que las moléculas MP2, MP6, MP9, MP11, MP13, MP14, MP19 Y MP20 fueron las más suaves indicando que presentan mayor reactividad que la cefotaxima cuyo valor de suavidad fue 0,227. Mientras que las moléculas MP1, MP3, MP4, MP5, MP7, MP8, MP10, MP12, MP15, MP16, MP17 y MP18 presentaron una dureza mayor que el valor de referencia calculado (2,203 eV), por lo que posiblemente presentarán menor reactividad con respecto a la cefotaxima.
El análisis de los mapas de potencial electrostático molecular permite inferir que estas moléculas tienen alta capacidad de formar interacciones electrostáticas con residuos de aminoácidos en el centro activo de las enzimas, favoreciendo acoplamiento ligando-receptor.
Los resultados obtenidos muestran que todas las moléculas presentaron mejores valores de afinidad por la enzima PBP, en comparación con el ligando nativo, mientras que 13 moléculas mostraron además menores valores de energía de unión por la enzima BLEE, de las cuales MP9 y MP16 presentaron el menor valor de energía de unión, por lo que se deduce dichas moléculas tienen posible resistencia a la enzima BLEE. Por último, se considera a las moléculas MP9, MP16 y MP17 como potenciales inhibidores de transpeptidasas y resistentes a BLEE producidas por E. coli, indicando que los grupos sustituyentes de estas moléculas son adecuados para el diseño de fármacos con mejor eficacia antibacteriana contra este microorganismo.


