











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El acceso a los medicamentos es un elemento esencial para el goce del grado máximo de salud; la Declaración universal de bioética y derechos humanos comprende esto como un derecho fundamental de todo ser humano, aclarando que "los progresos de la ciencia y la tecnología deberían fomentar el acceso a una atención médica de calidad y a los medicamentos esenciales [...] ya que la salud es esencial para la vida misma y debe considerarse un bien social y humano" [1]. Esta justificación debería ser suficiente para aceptar y estimular a los Estados a implementar políticas para asegurar el acceso a los medicamentos que se enmarca en un campo más amplio abarcando la promoción de salud, prevención y rehabilitación [2].
Haciendo un análisis retrospectivo, nos encontramos con el Acuerdo sobre los Derechos de Propiedad Intelectual relacionados con el Comercio (ADPIC) como uno de los anexos (1C) del Acuerdo de Marrakech de la Organización Mundial del Comercio (OMC), firmado en Marruecos, el 15 de abril de 1994. Lo concerniente a las patentes se encuentra en la parte II (Normas relativas a la existencia, alcance y ejercicio de los derechos de propiedad intelectual). En ella no se hace distinción entre los medicamentos o cualquier otro producto ya que "las patentes podrán obtenerse por todas las invenciones, sean de productos o de procedimientos, en todos los campos de la tecnología, siempre que sean nuevas, entrañen una actividad inventiva y sean susceptibles de aplicación industrial". Se establecen los derechos de exclusividad al titular de las patentes y se fija la duración de la protección en 20 años [3].
De otro lado, en respuesta a las necesidades de salud pública ya enunciadas, años más tarde surge la Declaración ministerial de Doha sobre el ADPIC y la salud pública, que es considerada por algunos autores como un avance sustancial en materia de acceso a los medicamentos. Esta declaración privilegia los intereses de la salud pública sobre los de propiedad industrial, al separar los productos farmacéuticos de los demás productos comerciales: "Reconocemos que la protección de la propiedad intelectual es importante para el desarrollo de nuevos medicamentos. Reconocemos asimismo las preocupaciones con respecto a sus efectos sobre los precios".
A la vez que insiste sobre la importancia de la flexibilidad de las normativas sobre las patentes destacando que "El acuerdo sobre los ADPIC no impide ni deberá impedir que los miembros adopten medidas para proteger la salud pública y, en particular, de promover el acceso a los medicamentos para todos" [4].
Actualmente, se sigue trabajando en quitar limitaciones a la producción, distribución y consumo de los medicamentos, considerando que son bienes que afectan la salud y pueden tener un importante impacto negativo, aunque se ha de preservar un delicado equilibrio con el alto ritmo de innovación propio del mercado de medicamentos.
METODOLOGÍA
Se ha realizado una revisión bibliográfica de artículos científicos y protocolos de uso habitual en medicamentos genéricos. Además, se consultaron los informes de la EMA y la FDA. Finalmente, se tuvo en cuenta un estudio comparado, las normas particulares que recogen las distintas legislaciones respecto a las normas de bioequivalencia y bioseguridad.
Hacia una definición de medicamentos genéricos
Según la Organización Mundial de la Salud (OMS), un medicamento genérico es aquel vendido bajo la denominación del principio activo que incorpora, siendo bioequivalente a la marca original. Para la OMS dos especialidades medicinales son bioequivalentes cuando siendo equivalentes o alternativas farmacéuticas sus biodisponibilidades después de la administración de la misma dosis molar son semejantes en tal grado que pueda esperarse que sus efectos sean esencialmente los mismos.
Los medicamentos genéricos tienen múltiples definiciones y dimensiones legales, farmacológicas, clínicas y económicas. En 1996 la OMS proponía la noción de "medicamentos de múltiples fuentes", también se mencionan como "medicamento competidor", "medicamento similar", "producto farmacéutico multiorigen", "medicamento intercambiable". Hoy en día podríamos concluir que un medicamento genérico es aquel fármaco que no es producido por el laboratorio innovador que desarrolló el principio activo, es decir, el laboratorio que lo produjo por primera vez y lo patentó [5]. Así, un medicamento genérico puede ser elaborado una vez vencida la patente del medicamento de marca siempre que reúna todas las condiciones de calidad y bioequivalencia (OMS-OPS).
Por su parte, la Unión Europea (UE) ha establecido que un producto farmacéutico genérico es un producto medicinal que tiene la misma composición cualitativa y cuantitativa en sustancia activa, la misma forma farmacéutica que el medicamento de referencia y cuya bioequivalencia haya sido demostrada por estudios de biodisponibilidad adecuados. En esta definición se asume que, en un mismo sujeto, un curso temporal de concentraciones plasmáticas similares resultará en concentraciones esencialmente iguales en el lugar de acción y, por tanto, con un efecto básicamente igual [6].
En Argentina, el Decreto 150/92 la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT) define el medicamento genérico como aquel principio activo o droga farmacéutica o, cuando corresponda, de una asociación o combinación de principios activos a dosis fijas, adoptada por la autoridad sanitaria nacional o, en su defecto, la denominación común internacional de un principio activo recomendada por la Organización Mundial de la Salud.
En resumen, el término y el concepto de medicamento genérico se utilizan de diversas maneras, pero está claro que su existencia sigue a la caducidad de una patente, y en ninguna definición se incluyen términos económicos de modo explícito.
El mercado de los genéricos
Los medicamentos genéricos están jugando un papel de primera magnitud, tanto cuantitativa como cualitativamente en la evolución del mercado farmacéutico. Este ha sido un tema controvertido por las variantes que han introducido este mercado y por los importantes intereses que se mueven alrededor del mismo. Es de gran relevancia la posición de los profesionales médicos, tanto en los aspectos científico-técnicos, como por su papel de interlocutores e intermediarios privilegiados entre el paciente y las administraciones sanitarias.
No se trata de desalentar la innovación, pero es muy importante entender que no toda la protección a la innovación debe llevarse a cabo desde el precio del medicamento ni la defensa de la innovación no puede basarse en sembrar dudas sobre la calidad, eficacia y seguridad de los medicamentos genéricos en su conjunto, que siguiendo las normativas de cada país no tendría por qué darse.
Las recomendaciones internacionales en políticas farmacéuticas establecen la necesidad de promover la competencia en la oferta de medicamentos como una importante estrategia para mejorar el acceso a los mismos. En la medida en que ingresen nuevos oferentes al mercado, la competencia por precios se fortalecerá y tenderán a converger en un commodity value. Esto significa que los precios de estos productos se homogeneizarían a nivel internacional con valores muy próximos a su costo de producción y distribución en el mercado, con la consecuente reducción de precios [7].
Sin embargo, la investigación biomédica -y en particular la que tiene que ver con nuevos fármacos- requiere de una gran inversión a lo largo de bastante tiempo y, consecuentemente, necesita de una protección suficiente que le permita amortizar la inversión realizada. De ahí, las políticas de patentes y de protección de datos que garantizan, durante un tiempo, la exclusividad en la explotación comercial.
Pero esta política debe acompañarse obligatoriament e de otra tendiente a reducir notablemente el precio de los medicamentos una vez que han vencido sus patentes y sus períodos de protección. Se puede asegurar que sin esta segunda política es absolutamente imposible mantener la primera; y es en esta política donde juega un papel primordial el medicamento genérico que es mucho más barato por la simple razón de que no tiene que amortizar las inversiones en investigación, parte más importante de los costes de un medicamento, que deben soportar no sólo los costos de los proyectos exitosos, sino también de los fracasos.
Los recursos de que disponemos son indudablemente escasos y nadie entendería que teniendo la posibilidad de acceder a un determinado tratamiento con un coste reducido, se optara por una alternativa más costosa siendo igual de efectiva, sobre todo si consideramos que ese exceso de recursos utilizado puede redundar en la imposibilidad de llevar a cabo otros tratamientos o dar cobertura a otras patologías o pacientes.
Por su parte, el CEO de Merck aseguraba que
El precio de los medicamentos no se fija según los costos de investigación. Al contrario, se fija según su valor para prevenir y curar las enfermedades. Merck puede gastar quinientos o mil millones de dólares en el desarrollo de un medicamento, pero son el médico, el paciente y los que pagan por nuestras medicinas los que fijan su verdadero valor [8].
Así, se desea establecer un control en la formación del precio de venta al público de los medicamentos, limitando los márgenes de comercialización (techo para las ganancias de farmacias y droguerías). Lo que implica controlar los márgenes de farmacia de los distintos actores que intervienen en la cadena de producción y comercialización [9].
En la tabla 1 puede observarse el nivel de aceptación de un cambio de medicación de marca a genérico frente una mejora en el precio. La muestra poblacional corresponde a Barcelona, España.
Tabla 1 Porcentaje de pacientes que aceptaron el cambio a genérico en función del tipo de fármaco.
| Número de pacientes | Porcentaje de aceptación | |
|---|---|---|
| Analgésicos/AINE a | 137 | 100,0 |
| Antineoplásicos | 57 | 100,0 |
| Digestivo | 580 | 99,3 |
| Cardiovascular | 2406 | 99,1 |
| SNC a | 767 | 97,7 |
| Otros b | 673 | 99,0 |
Fuente: versión propia con base la Ref. [10]. a AINE: antiinflamatorio no esteroideo; SNC: sistema nervioso central. b Flutamida, tamoxifeno, acetilcisteína, alopurinol, gemfibrozilo, ticlopidina, timolol, aciclovir, cipro-floxacino y norfloxacino.
Regulaciones de mercado
La tesis clásica de la mano invisible de Adam Smith ha dejado paso a la necesidad de regular los mercados, especialmente en una industria tan sensible como la farmacéutica, donde no se dan los supuestos de la competencia perfecta. Se observa una creciente tensión en la normativa internacional que regula el derecho de propiedad intelectual impulsado en el ADPIC, de la Organización Mundial del Comercio (OMC), y el derecho a la salud contemplado en el pacto internacional de derechos económicos, sociales y culturales (PIDESC) de la Organización de las Naciones Unidas [11].
En Estados Unidos, la enmienda conocida como Hatch-Waxman Act (Drug Price Competition and Patent Restoration Act) que estimula el desarrollo de productos genéricos estableció en 1984 un sistema de aprobación abreviado para introducir "nuevas versiones comerciales" de un medicamento ya aprobado.
La ley de Hatch-Waxman fue concebida para inyectar competencia al mercado de medicamentos de venta bajo receta, tratando de mantener los derechos de propiedad intelectual de los inventores/descubridores del producto farmacéutico. Desde su promulgación, se ha demostrado que los precios de los productos genéricos disminuyen en relación directa al número de genéricos que entran al mercado. Con anterioridad a la promulgación de esta ley, los productores de genéricos debían llevar a cabo exactamente los mismos estudios de seguridad y eficacia que los inventores originales para registrar y comercializar el producto.
Esto implicaba un factor inhibidor, para las compañías de productos genéricos, pues los costos eran prohibitivos. La ley Hatch-Waxman permitió disminuir el costo del desarrollo de los productos genéricos mediante una simplificación y abreviación del proceso, pero siempre requiriendo demostrar la equivalencia del genérico al producto de referencia, bajo rigurosas reglas basadas en principios científicos bien desarrollados. Además, estipuló que el proceso de manufactura del medicamento debía de adherirse a todas las regulaciones aprobadas por la FDA, y permitió a las compañías que producen genéricos, empezar sus actividades de desarrollo antes de que expire la patente del producto original [12].
Si bien la ley Hatch-Waxman tuvo un efecto importante en disminuir el costo de los medicamentos, a su vez generó una serie de respuestas de la industria farmacéutica para aminorar ese impacto, utilizando otros mecanismos para retrasar el ingreso de genéricos tales como: (a) patentar todo lo patentable en un producto al punto que cada medicamento de marca hoy en día tiene alrededor de diez patentes; (b) hacer acuerdos exclusivos con compañías que producen insumos, para limitar el acceso a estos insumos; (c) cambiar las fórmulas farmacéuticas y retirar del mercado la forma farmacéutica original; (d) lanzar su propio producto genérico al mercado, de acuerdo con una compañía de genéricos, para seguir controlando el mercado al menos los primeros 180 días de comercialización; (e) establecer acuerdos de no comercialización con la compañía de genéricos que recibió la aprobación; (f) tratar de obtener licencia del producto para venta sin receta médica (Over the counter: OTC) [13].
Los medicamentos genéricos en Europa y Estados Unidos
La difusión de los medicamentos genéricos en España ha sido posterior a su introducción en otros países del centro y norte de Europa (Alemania, Países Bajos y Escandinavia) con mayores cuotas de mercado de medicamentos genéricos, en donde esta implantación se produjo ya en la década de los 80, motivo por el que la penetración y cultura de utilización de estos medicamentos es muy superior a países como Francia, España, Portugal y Grecia, donde las medidas de promoción de la utilización de estos medicamentos se remontan a finales de la década de los 90 [14].
Los países pertenecientes a la Unión Europea (UE) han desarrollado, a través de la Agencia Europea del Medicamento, una homogeneización en materia de autorización de medicamentos y exclusividad de los datos. En la UE el registro de medicamentos de uso humano se rige por la Directiva 2004/27/CE del Parlamento Europeo y del Consejo de 31 de marzo de 2004, que modifica la Directiva 2001/83/CE, por la que se establece un código comunitario sobre medicamentos de uso humano, y por el Reglamento (CE) 726/2004 del Parlamento Europeo y del Consejo de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea del Medicamento.
Muchos países han adoptado exenciones jurídicas (o exenciones con fines de investigación) de infracción respecto de determinados actos relacionados con el desarrollo y la presentación de datos de ensayos ante organismos reguladores. Esas exenciones suelen recibir el nombre de disposiciones «Bolar», en alusión a la ley estadounidense por la que se derogaba un fallo judicial en el que se confirmaba que los Estados Unidos de América no contemplaban una exención con fines de investigación (Roche Products, Inc. contra Bolar Pharmaceutical Co., Inc., 733 F.2d 858, 1984).
Así, se introduce la «disposición Bolar» que habilita a las empresas farmacéuticas a realizar el desarrollo, las pruebas y la experimentación necesarios para conseguir el registro de un medicamento genérico antes de que finalice el período de protección de la patente del medicamento innovador, sin violar por ello la reglamentación relativa a la protección de la propiedad industrial y comercial. Esta disposición, que no estaba contemplada en la anterior redacción de la Directiva 2001/83/CE, permite que los medicamentos genéricos puedan estar a disposición de los pacientes inmediatamente después del vencimiento de la patente y del período de protección de datos del medicamento innovador, además de evitar que una gran parte de las pruebas requeridas se efectúen en terceros países en lugar de en la UE.
En Estados Unidos, antes de que se aprobase la ley Hatch-Waxman en 1984, los productores de genéricos tenían que repetir las pruebas de seguridad y eficacia que había hecho la compañía innovadora. La ley permitió que los productores de genéricos utilizaran e hicieran referencia a la investigación existente cuando presentaran una solicitud a la Food and Drug Administration (FDA) para producir un genérico y obligaba a la compañía innovadora a compartir la información sobre las investigaciones que se habían hecho, con los productores de genéricos antes de que expirase la patente para que la compañía de genéricos pudiera probar su propia producción.
La Oficina de Fármacos Genéricos del CDER (Office of Generic Drugs, OGD), perteneciente al Centro para la Evaluación e Investigación de Fármacos (CDER por sus siglas en inglés), es el organismo encargado de garantizar que los medicamentos genéricos sean seguros, eficaces y bioequivalentes con el producto de referencia.
Es importante aclarar que la FDA asume la equivalencia terapéutica cuando dos preparados son equivalentes farmacéuticos tienen el mismo efecto terapéutico, son bioequivalentes y han sido fabricados cumpliendo sus normas vigentes de buena práctica de fabricación. En agosto de 2000, el Centro para la Evaluación de Medicamentos e Investigación (CDER) de la FDA publicó un documento denominado Dispensa de estudios de biodisponibilidad y bioequivalencia in vivo para formas farmacéuticas orales sólidas de liberación inmediata, aplicable en general para modificaciones de productos ya registrados [15]. Estas dispensas o excepciones se basan en la clasificación biofarmacéutica que estratifica a los principios activos en cuatro categorías, de acuerdo con su solubilidad y absorción:
Clase 1: alta solubilidad y alta permeabilidad.
Clase 2: baja solubilidad y alta permeabilidad.
Clase 3: alta solubilidad y baja permeabilidad.
Clase 4: baja solubilidad y baja permeabilidad.
La demostración de bioequivalencia puede no ser necesaria para medicamentos que contienen principios activos en la clase 1 y algunos de clase 3.
Los estudios de bioequivalencia realizados para la aprobación de las drogas genéricas son similares a los requeridos en Europa y se basan en los mismos parámetros farmacocinéticas: concentración máxima (Cmax), tiempo máximo (Tmax) y área bajo la curva (area under the curve, AUC). Asimismo, se consideran bioequivalentes aquellos medicamentos en los que con un intervalo de confianza del 90% la razón entre las medias de los parámetros investigados está dentro del límite 80-125% para el parámetro principal AUC y para la Cmax [16]
Los criterios de bioequivalencia bien aceptados, AUC y Cmax son relativamente empíricos. Los estándares de disolución de medicamentos, por otro lado, se pueden establecer sobre una base mecanicista. Si dos productos farmacéuticos tienen el mismo perfil de disolución in vivo en todas las condiciones luminales tendrán la misma velocidad y grado de absorción, y serán bioequivalentes [17].
En América Latina se observan tres tendencias regulatorias, la primera que favorece la difusión de medicamentos competidores, la promoción extendida de uso de medicamentos genéricos, y no ponen límites a la sustitución de medicamentos innovadores por competidores; quienes, si ponen límites a esa competencia, y aquellos países que aún tienen un desarrollo incipiente.
En Argentina, Colombia y Brasil, la financiación de medicamentos otorga ventaja a los competidores o genéricos. En Ecuador, el sistema de salud no es de cobertura universal, sin embargo, la provisión de medicamentos competidores es obligatoria [18].
Aunque las normas de propiedad intelectual en el campo farmacéutico se aplican en los Estados Unidos y algunos países europeos desde hace más de 50 años, su vigencia en América Latina, el Caribe y la mayoría de los países en desarrollo comenzó en 1995, después de la creación de la Organización Mundial del Comercio (OMC) y la adopción del Acuerdo sobre los aspectos de los derechos de propiedad intelectual relacionado con el comercio [18].
La aplicación de las leyes de mercado a los fármacos
El mercado de medicamentos genéricos se caracteriza por una alta dispersión de precios. No hay ningún otro bien de consumo en el mercado mundial con tanta diferencia de precios entre diversos productos que responden a las mismas especificaciones técnicas. Asimismo, presenta inelasticidad de los precios; la demanda de medicamentos no varía proporcionalmente con las variaciones de precio.
En una situación de libre mercado y en presencia de una industria farmacéutica altamente concentrada, esta inelasticidad deja al consumidor en una situación de gran vulnerabilidad ante los aumentos de precio, siendo regresiva la financiación. Los pobres gastan en medicamentos relativamente más que los ricos, porque tienen mayores necesidades que los ricos y disponen de menos condiciones para conseguirlos. En los países en vías de desarrollo, una porción mayor de la financiación de los medicamentos depende de los ingresos de los hogares, es decir, el gasto en medicamentos en los países desarrollados se financia predominantemente con recursos públicos, mientras que en los países en desarrollo se financia mayormente con recursos privados [7].
Evolución del mercado de genéricos
Existen algunos datos sobre la evolución de medicamentos genéricos que difiere de país en país, aunque la tendencia a nivel mundial es creciente.
Por ejemplo, en el mercado español hasta 2014 las marcas venían evolucionando negativamente y los genéricos de modo positivo. En el año 2015 se observa un cambio en la tendencia, que se mantiene en el año 2016. Según datos del Ministerio de Salud, el consumo de los genéricos con receta representa un 48,8 % del consumo total, y un 20 % en valores facturados [19]. Encontrándose próxima a la media de consumo de genéricos entre los países de la Organización para la Cooperación y el Desarrollo Económico (OCDE), según las estadísticas publicadas por ese organismo.
La Asociación Española de Medicamentos Genéricos (AESEG) señala que las cuotas de medicamentos genéricos (tanto en volumen como en valor) están alejadas de los países de referencia europeos, Alemania y Reino Unido, con un volumen de genéricos cercanos al 80% del total de mercado farmacéutico y con un valor de alrededor del 35% de todo el gasto farmacéutico. La misma fuente señala que el ahorro anual generado por los medicamentos genéricos en el Reino Unido asciende a los seis mil millones de euros.
Respecto a los datos correspondientes a 2012, el consumo medio de genéricos en los 23 países analizados de la organización se situaría en el 45% de las unidades consumidas, y se correspondería con el 23% del valor económico total del mercado farmacéutico.
El país de la OCDE que más genéricos consume es Estados Unidos, el 84% de lo dispensado en la oficina de farmacia pertenece a esta categoría, aunque sólo correspondería al 28% de la factura farmacéutica. Seguido por Chile, con el 81% de las unidades. En este país, sin embargo, el valor económico de los medicamentos genéricos se elevaría hasta el 59% del total, el más alto entre las naciones. Reino Unido y Alemania también estarían entre los primeros puestos, con niveles de consumo superiores a las tres cuartas partes de su mercado total. De los cinco principales mercados farmacéuticos europeos, España se sitúa justo en el medio, pues Francia (27%) e Italia (18%) registran cifras menores de uso.
De otro lado, encuestas llevadas a cabo entre 2007 y 2012 indicaron que en los países de ingresos bajos y medianos determinados medicamentos genéricos sólo estaban disponibles en el 56% de los puntos públicos de distribución de medicamentos. Los precios que los pacientes debían pagar en el sector privado por los medicamentos genéricos más baratos eran, en promedio, 5 veces superiores a los precios de referencia internacionales, y en algunos países llegaban a ser unas 14 veces más altas. En consecuencia, incluso los tratamientos basados en los genéricos de menor precio resultan aún inaccesibles para muchos hogares con ingresos bajos. El problema se agrava cuando varios miembros de una misma familia enferman al mismo tiempo [20].
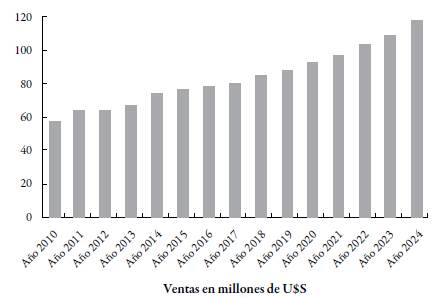
Fuente: Ref. [21].
Figura 1 Evolución anual del valor de las ventas de medicamentos genéricos con receta a nivel global de 2010 a 2024 (en millones de dólares).
Ley de medicamentos genéricos en Argentina
El 28 de agosto de 2002 se sancionó en Argentina la Ley 25.649 de especialidades medicinales y promoción de la utilización de medicamentos por su nombre genérico, que indica que toda receta o prescripción médica deberá efectuarse en forma obligatoria expresando el nombre genérico del medicamento. Sin embargo, en el segundo párrafo de la ley se puntualiza como criterio adicional que la receta podrá indicar, además del nombre genérico, el nombre o marca comercial. Este agregado dejó abierta la puerta para que, luego de un período de buenos resultados (hasta el año 2006), comenzara el incumplimiento de la ley.
Por su parte, el organismo regulador de medicamentos (ANMAT) sostiene que no todos los medicamentos requieren pruebas de bioequivalencia para poner en práctica la sustitución genérica. Para ello cuentan con un programa de Bioequivalencia que clasifica los medicamentos que requieren esta prueba sobre la base de la importancia de los efectos que pueden ocasionar en concentraciones fuera de la llamada "ventana terapéutica"; a esto lo denominan "riesgo sanitario". Este criterio se basa en las categorías de las drogas clasificadas en el sistema de clasificación biofarmacéutica (SCB).
Los principios activos bioexceptuados son los de clase I: alta solubilidad y permeabilidad y algunos de clase III: alta solubilidad y baja permeabilidad. Estos principios activos incluidos dentro de la categoría de riesgo intermedio y bajo están listados en los documentos de la OMS [22]. La Organización Panamericana de Salud promueve los procesos de armonización en las américas a través de la Red Panamericana para la Armonización y Regulación Farmacéutica (REDPARF) que agrupa diferentes instancias de armonización en la región (Grupo Andino, Caricom, Mercosur, Nafta, Sistema de Integración Centroamericana) así como agencias reguladoras, académicos y consumidores. Dentro de esta red se creó el Grupo de Trabajo de Bioequivalencia y Biodisponibilidad en noviembre de 1999 [23].
En contraste con los países europeos, en Latinoamérica los medicamentos genéricos han logrado una baja incorporación dentro de los mercados [24].
DISCUSIÓN
Es una realidad que el precio es una de las principales barreras de acceso a los medicamentos. El gasto en todas las actividades de I+D, incluyendo tanto el de los proyectos que terminan con un producto más o menos exitoso en el mercado, como el de los proyectos fallidos en cualquier fase del proceso de I+D, deben ser recuperados a través de las ventas de los productos que tienen éxito comercial.
Frente a esta realidad se encuentran los sistemas de salud y los organismos internacionales que propenden por el acceso equitativo y a tiempo de los medicamentos por parte de la población mundial, de un modo especial la de los países en vías de desarrollo que padecen de un modo especial las asimetrías del mercado farmacéutico.
Se establece así necesidad de revisar una vez más la ley de protección de patentes, de modo que sin desalentar el desarrollo de I+D, permita un mejor acceso de los medicamentos, en muchos casos estamos ante enfermedades graves que pueden volverse mortales por falta de tratamiento adecuado a tiempo. Sin dejar de mirar de los países desarrollados, es necesaria una consideración más humanitaria hacia el continente africano y América Latina, donde la disponibilidad de productos farmacéuticos genéricos y baratos es la única fuente de acceso a una medicina asequible por parte de millones de personas.
El precio es una de las principales barreras de acceso a los medicamentos, como ya se ha mencionado, y hacen peligrar la sostenibilidad de los sistemas de salud. Por ello es necesario diseñar formas de intervención pública que puedan modificarlos para hacerlos asequibles, manteniendo un nivel adecuado de innovación. Una solución satisfactoria a largo plazo requeriría una acción coordinada de los compradores a nivel internacional, así a la oferta global que suponen las empresas multinacionales se contrapone una demanda global de países organizados o coordinados [25], y, por otro lado, un cambio sustancial en el actual sistema de patentes.


