











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
El descubrimiento de candidatos químicos es la fase primaria del desarrollo de nuevos tratamientos farmacológicos. Consiste en la búsqueda de nuevas moléculas capaces de generar respuesta diferenciada en la actividad de un modulador de enfermedad (elemento diana capaz de regular una red biomolecular importante en la progresión de una enfermedad) [1]. Este proceso de búsqueda es denominado investigación básica y comprende el inicio de la fase de descubrimiento de fármacos.
Durante la investigación básica pueden ser descritas y caracterizadas dianas terapéuticas clave en la generación de un estado de equilibrio entre un estado normal y un estado de pre enfermedad, sensible a estímulos externos. Estos comprenden dos de los tres estados de progresión de la enfermedad, donde en el último estado (estado de enfermedad) existe un deterioro grave [2]. Por lo tanto, es importante garantizar que la diana sea modulable (druggable) [3], lo cual significa que, al interactuar con un compuesto químico o una proteína, logrará disparar una respuesta biológica que altera el curso de la enfermedad retornando al estado normal.
Algunos estudios logran este cometido gracias a su enfoque en la regulación fenotípica de ese estado de equilibrio y posterior validación de sus resultados [4]. Estos últimos establecen la base de trabajo para las subfases del descubrimiento de fármacos (chequeo primario, secundario y ensayos farmacocinéticos). El chequeo primario o (Primary screening) evalúa la capacidad de una librería de compuestos para afectar o modular la función de la diana terapéutica. Posteriormente se determina la relación cantidad-respuesta de estos compuestos y los mecanismos moleculares responsables de su actividad; esto se denomina chequeo secundario o (Secondary screening). Los resultados originados en este punto permiten el rediseño de la estructura molecular del compuesto con el fin de optimizar su respuesta. Posterior a estas subfases, se evalúa la farmacocinética de la molécula (absorción, distribución, metabolismo, excreción y toxicidad) en modelos fisiológicos [5]. De este modo se asegura la idoneidad de la molécula antes de proseguir con la fase de desarrollo de fármacos (Investigación preclínica, ensayos clínicos, eficacia a pequeña escala; eficacia a gran escala y aplicación clínica) [6-8].
La fase de descubrimiento gira entorno a la interacción entre una molécula y una diana modulable. Para que una molécula sea seleccionada como compuesto candidato, deben ser caracterizadas sus diferentes propiedades derivadas de la interacción molécula-entorno [9]. Estas interacciones pueden depender de la estructura molecular; por lo cual, la diana terapéutica debe afectar de manera significativa y específica el sistema de equilibrio entre estados de la enfermedad, permitiendo así dirigir adecuadamente el diseño o rediseño de la estructura molecular.
La estructura molecular determina las propiedades fisicoquímicas, bioquímicas, farmacocinéticas y toxicológicas de un compuesto. Así, cuando una molécula interactúa con su entorno, observamos interacciones determinadas por sus propiedades estructurales (capacidad de donar o aceptar electrones, capacidad de interacción por puentes de hidrógeno, lipofilicidad, peso o tamaño molecular, polaridad, etcétera) [10]. Estas interacciones se denominan propiedades fisicoquímicas (área de superficie polar, lipofilicidad, permeabilidad, reactividad, solubilidad, etcétera) cuando la naturaleza de su entorno es física [11]. Propiedades bioquímicas cuando la naturaleza de su entorno es biológica (absorción, metabolismo, unión a proteínas y tejidos, etcétera) [12]. La interacción entre estas últimas y sistemas vivos, determinan las propiedades farmacocinéticas y toxicológicas (eliminación, biodisponibilidad, dosis letal 50, toxicidad idiosincrásica, vida media, etcétera) [13]. Por lo tanto, la estructura de una molécula es un factor determinante en el descubrimiento de nuevos candidatos químicos.
METODOLOGÍA
Búsqueda bibliográfica y fuentes de información
Se realizó una búsqueda de información relacionada con investigación biomédica y el descubrimiento de fármacos. Los estudios se identificaron a través del motor de búsqueda de Google, Google Scholar, el cual indexa editoriales, bibliotecas, repositorios, bases de datos bibliográficas, entre otros. Se tuvieron en cuenta listas de referencias de artículos y consulta con un experto para la realización de la búsqueda de información.
La estrategia de búsqueda se compuso de términos claves y operadores booleanos (AND) para optimizar la búsqueda. Los términos utilizados fueron:
Biomedical Research.
Complex diseases AND Biomolecular networks.
Drug discovery AND Drug development.
Live cells AND Drug development.
Live cells AND Click chemistry.
Live cells AND Proteomics.
CRISPR Cas9 author: Mojica.
Chemical ecology AND Network.
Molecular structure AND Drug development AND Live cell. Drug discovery AND Computational mapping. Drug discovery AND Molecular dynamic.
Drug discovery AND High throughput assay AND Live cell imaging. High throughput assay AND Live cell imaging AND Ex vivo.
Selección de los estudios
Mediante una lectura crítica y detallada de los títulos y resúmenes de los artículos, el investigador principal verificó que los términos claves, objetivos, metodología, resultados y conclusiones fueran relevantes, o que describieron el fenómeno de interés, para el cumplimiento de los objetivos de la presente revisión, al igual que fueran publicados en revistas reconocidas del tema.
Resultados de la selección de los estudios
La estrategia de búsqueda identificó 289 artículos, los cuales fueron revisados que cumplieran con todos los criterios de elegibilidad. Posteriormente, se hizo la selección para evaluar si el contenido desarrollado era pertinente, obteniéndose así un total de 51 artículos que fueron sometidos a un análisis profundo de su información, de los cuales un total de 36 son artículos de revisión y 15 son artículos originales. El investigador principal extrajo la información de los estudios incluidos, que posteriormente fue revisada por otro investigador. Además, se incluyeron 3 libros y 4 artículos fuera de la ventana de búsqueda para explicar conceptos y ampliar la información del tema. Por recomendación de un experto, se incluyó un video conferencia y 3 artículos originales para complementar la revisión (figura 1).
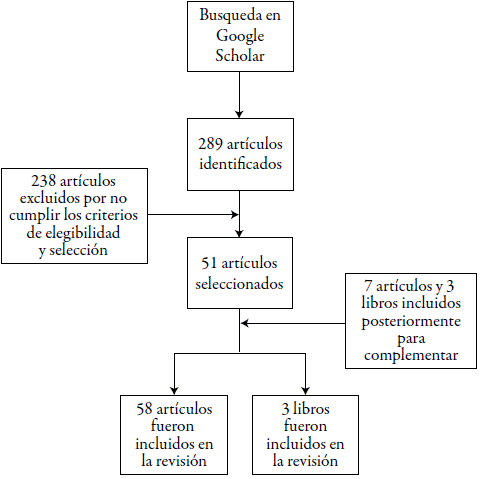
La brecha entre el descubrimiento y desarrollo de fármacos
El conocimiento sobre dianas terapéuticas obtenido de la investigación básica debe permitir superar la estrecha brecha de translación que separa el descubrimiento de fármacos del inicio del desarrollo preclínico [14]. Esta brecha puede presentar diferentes problemáticas limitantes, posiblemente relacionadas con la interacción entre candidato químico y diana en un entorno celular. Esas problemáticas se denominan bloqueo de la innovación [15, 16]. De este modo, consideramos necesario integrar la identificación de potenciales dianas con una adecuada caracterización de compuestos teniendo los mecanismos patogénicos de la enfermedad; para posteriormente observar y monitorear una respuesta en el comportamiento celular.
El bloqueo de la innovación entre las fases de descubrimiento y desarrollo de fármacos debe ser resuelto. Más adelante describiremos cómo la interacción candidato químico-diana terapéutica, evaluada durante los procesos de caracterización e identificación, puede ser abordada desde la captura de imágenes de células vivas permitiendo solucionar esa problemática de translación.
La estructura molecular y su importancia en el descubrimiento de fármacos
Ante la diversidad estructural de las moléculas existentes en la naturaleza y sintetizadas por el hombre [17-19], se observa una limitada disponibilidad de compuestos a evaluar frente al creciente panorama de dianas terapéuticas. Esto se debe a procesos naturales de interacción metabólica entre especies de organismos estudiada por la ecología química (como por ejemplo la facilitación, mutualismo, relación huésped-parásito, alelopatía y relación presa-predador) [20-22]. Esto ofrece un panorama más reducido en la búsqueda de nuevos fármacos; por lo cual, las librerías de compuestos no proporcionan alta diversidad estructural comparada con los productos naturales. Los pocos resultados destacables en el descubrimiento de fármacos son su evidencia [23]. En este sentido, la búsqueda de nuevas moléculas debe realizarse desde fuentes naturales y teniendo en cuenta su relevancia en sistemas biológicos.
Los complejos mecanismos celulares pueden ser aproximados a diferentes aspectos de la biología de las enfermedades [24]. Partiendo de este patrón, es posible optimizar eficientemente la caracterización de moléculas como posibles candidatos químicos, teniendo en cuenta los cambios fenotípicos en líneas celulares específicas [9]. Además, las interacciones de los compuestos con un entorno celular podrían dirigir la búsqueda hacia una diana concreta; evitando así actividad indeseada en otros receptores distintos a la diana terapéutica [25]. Aquí proponemos la imagenología de células vivas y la modelación matemática de redes de regulación de genes y vías de señalización como herramienta de monitorización cuantitativa del comportamiento celular ante un compuesto [26].
Caracterizar una molécula como posible compuesto candidato capaz de generar cambios fenotípicos en un sistema biológico, involucra consideraciones estructurales adecuadas a partir de los diferentes factores que definen las propiedades de interacción molécula-entorno. Estas interacciones son fundamentales para otorgar actividad y selectividad al compuesto [27]. Garantizar un equilibrio específico entre propiedades fisicoquímicas y bioquímicas proporciona cierto nivel de interacción con el blanco terapéutico, tal que se logre la modulación requerida. Además, el compuesto de presentar buena solubilidad acuosa, estabilidad metabólica y permeabilidad [28].
Es posible implementar el diseño estructural computacional de compuestos como herramienta para determinar dicho equilibrio de interacción [23, 29]. Esta herramienta predice la energía libre de unión a partir de la flexibilidad estructural entre el compuesto candidato y su diana cuando se da una interacción entre ambos. Esta predicción resulta de simulaciones, basadas en la dinámica molecular clásica, donde se identifican trayectorias espaciotemporales (posición y velocidad) de los átomos involucrados en la interacción [23, 30]. De este modo, la selección o diseño estructural puede ser enfocado en una interacción específica; siempre y cuando se tenga en cuenta parámetros estructurales generales adecuados para el equilibrio de interacción.
Dentro de las consideraciones estructurales se encuentra que el tamaño molecular puede determinar el grado de interacción entre el candidato y el blanco [31]. Compuestos pequeños, con peso molecular <500 Da o menos de 20 átomos sin contar átomos de hidrógeno [32], presentan enlaces C-H que aportan significativamente a la energía libre de unión [30]; siempre y cuando estos enlaces pertenezcan a los grupos funcionales más próximos a la superficie del blanco terapéutico [29]; por lo tanto, a mayor energía libre de unión, mayor afinidad de unión entre candidato-blanco.
El área de superficie polar, medida en Ao2, es importante para la predicción de permeabilidad y solubilidad a partir de la polaridad; esta es asociada a la formación de puentes de hidrógeno para valores entre 50 y 100 Ao2, los cuales también contribuyen a la energía libre de unión [29, 31]. Sin embargo, esta propiedad fisicoquímica no considera átomos electronegativos diferentes a oxígeno y nitrógeno; los cuales también aportan a la polaridad global del compuesto [33]. Una estrategia útil para predecir la polaridad de manera más acertada es combinar el cálculo del área de superficie polar con descriptores numéricos cuantitativos como VolSurf. Estos descriptores son extraídos de mapas de campos moleculares (calculados a partir de la configuración espacial de la molécula) [33, 34].
La lipofilicidad, expresada como logaritmo del coeficiente de partición entre fases inmiscibles (P), está asociada a baja solubilidad acuosa (baja absorción) y aumento de carga metabólica para ser eliminado de circulación sistémica cuando presenta valores de logP >2 [35], generando toxicidad y por tanto fracaso del compuesto como candidato. El aumento de la carga metabólica puede generar metabolitos potencialmente reactivos, los cuales pueden presentar unión covalente a proteínas del metabolismo hepático (toxicidad idiosincrásica) [36-38]. También pueden considerarse los puentes de hidrógeno como predictores de coeficientes de partición alcano/agua (específico según la lipofilicidad del compuesto) [39]. Esto se debe a que donadores y aceptores de puentes de hidrógeno, presentes en la estructura de una molécula, favorecen la solvatación acuosa haciendo el soluto menos lipofílico.
Una vez caracterizados los posibles candidatos, éstos pueden ser evaluados por análisis de relación estructura actividad para identificar o validar los grupos funcionales que aumentan la interacción con el blanco [29]. Este esquema de estudio está arrojando información relevante en para el descubrimiento de fármacos [40]. Lo anterior demuestra que realizar modificaciones estructurales favoreciendo el equilibrio de interacción, aumenta la afinidad de un candidato por un blanco terapéutico. Sin embargo, para lograr modificar el curso de la enfermedad es necesario identificar correctamente el blanco terapéutico.
La complejidad biológica de las enfermedades en la identificación de dianas terapéuticas
La identificación de blancos relevantes para regular un proceso biológico es un paso indispensable en el descubrimiento de candidatos químicos. Actualmente se desarrollan e implementan estrategias que direccionan la búsqueda de dianas hacia enfoques basados en mecanismos específicos de procesos patológicos a partir de sistemas celulares [9, 24, 25]. Estas estrategias se agrupan en:
Enfoques basados en afinidad. Identifican proteínas capaces de interaccionar con un compuesto específico, para después clasificar sus interacciones según su funcionalidad [41]. Por ejemplo, la quimioproteómica utiliza sondas de afinidad, por enlace covalente, desarrolladas a partir del compuesto evaluado frente a líneas celulares específicas posteriormente lisadas para análisis proteómico (figura 2) [42-44]. La sonda está constituida por el compuesto de interés conjugado con un grupo reactivo y un grupo identificador de afinidad (figura 2A) [42]. En la sonda, el grupo reactivo interacciona de manera irreversible por oxidación directa o unión covalente con una región del sitio activo de la proteína (figura 2C). Un ejemplo de interacción irreversible es el caso de la sulfonación de la cisteína [45, 46] o el fluoruro de sulfonilo con lisina en el sitio de unión a ATP en pretinas tipo quinasas [47]. El grupo identificador de afinidad permite la diferenciación y cuantificación de proteínas interactuantes por captura de imágenes de células vivas. Generalmente son usados grupos alquilo para realizar química de "clic" con etiquetas fluorescentes [48].
Es importante identificar las regiones adecuadas del compuesto de interés donde pueda adicionarse el grupo reactivo y el grupo identificador de afinidad sin interferir con la actividad biológica de la molécula [28]. La síntesis de una sonda de afinidad puede ser llevada a cabo usando (Late stage functionalization) (LSF) como estrategia. Esta herramienta de síntesis considera la manipulación de enlaces Carbono-Hidrógeno del compuesto [28, 49]. Para esto, hace uso de reacciones químicas específicas según las características de la región molecular a modificar [50-53]. La implementación de LSF consiste en: 1. Identificar los enlaces carbono-hidrógeno a intervenir (facilitado por estudios de relación estructura actividad o modelamiento molecular computarizado) [49]. 2. Establecer las posibles reacciones realizables sobre los enlaces identificados, teniendo en cuenta patrones de selectividad por densidad electrónica. 3. Definir cuáles serán los grupos funcionales por insertar. 4. Prever el equilibrio entre propiedades fisicoquímicas y bioquímicas de las nuevas moléculas [28].
Este tipo de enfoque puede revelar una serie de dianas clave y garantizar el panorama proteómico específico de una enfermedad siempre y cuando se implemente en sistemas celulares definidos para el estudio de esa patología. Aquellas dianas identificadas presentarían cierto grado de interacción con la molécula usada en la sonda. Posteriormente, la molécula continuaría con el (Secondary screening) y estudios in vitro.
Enfoques basados en sistemas ex vivo. Estos sistemas son células o tejidos obtenidos directamente de un individuo. Son cultivados para representar un panorama clínico real, el contexto tisular normal o enfermo, el perfil genómico una enfermedad, el metaboloma específico del tejido, entre otras características [54]. Las líneas celulares convencionales no representan con precisión el comportamiento celular ex vivo de las enfermedades ante el tratamiento con algún compuesto [55]. Además, las líneas celulares convencionales pueden no representar genotípica o fenotípicamente una enfermedad debido su variabilidad genética respecto al tejido origen y a la influencia de condiciones de cultivo distintas a condiciones fisiológicas [54]. Lo anterior justificaría este enfoque como alternativa.
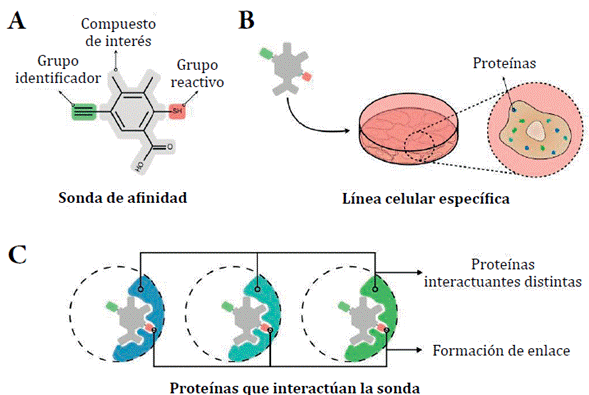
Figura 2 Proceso de interacción entre proteínas de interés y sonda de afinidad. A. Estructura general de la sonda de afinidad. Compuesta por un grupo identificador (verde), compuesto de interés (gris) y un grupo reactivo (rojo). B-C. Evaluación de la sonda en una línea celular específica. La sonda puede interactuar covalentemente con varias proteínas. Posterior análisis proteómico permite la identificación del perfil proteómico específico para dicha interacción.
El aislamiento del material cultivable como sistema ex vivo se realiza a partir de tejido relevante según la enfermedad o fenómeno que será estudiado. Este material es sometido a un proceso de mantenimiento (protocolos definidos según el tipo de tejido), el cual debe garantizar su viabilidad [56,-,58]. Este tipo de enfoque ha permitido: 1. Llevar a cabo ensayos de alto rendimiento utilizando hepatocitos primarios de ratones moduladores de esteatosis hepática; dando como resultado una tasa de éxito del 3,3% en el cribado de compuestos candidatos, así como la identificación de la Aurora quinasa C como blanco de inhibición [56]. Este enfoque también ha permitido la identificación de respuesta a tratamiento anticancerígeno para carcinoma de pulmón, utilizando cultivos de explantes del carcinoma [57]. Así como la predicción de la toxicidad asociada a moléculas específicas, como es el caso de la predicción de hepatotoxicidad del acetaminofeno usando cultivos ex vivo de tejido hepático humano [58].
Utilizar este tipo de enfoque ofrece ventajas referentes a la manera de intervenir moduladores capaces de alterar el equilibrio entre un estado normal y un estado pre-enfermedad. Sin embargo, puede ser difícil establecer un protocolo que garantice la viabilidad del cultivo en un tiempo adecuado para la captura de imágenes en células vivas. Esto, debido a los altos requerimientos de suplementos y medios. A raíz de esta dificultad, ha surgido la edición del genoma como un enfoque alternativo que busca simular las distintas rutas moleculares que intervienen en el equilibrio de estados de la enfermedad.
Enfoques basados en la edición del genoma. Este tipo de enfoque ha sido potenciado debido a la implementación de la tecnología CRISPR-Cas9. Esta metodología, ha permitido la edición precisa del genoma [54], gracias al desarrollo biotecnológico de sus componentes moleculares [59]. El sistema CRISPR-Cas9 ofrece una alternativa para el desarrollo eficiente de líneas celulares. Para lograr este propósito, se llevan a cabo modificación de genes específicos usando una secuencia guía de ARN (gARN) y una endonucleasa asociada a CRISPR tipo II (Cas9) [60]. El complejo riboproteico formado entre estos dos componentes se une a la secuencia ADN específica para el gARN, cortando la doble cadena de ADN para dicha secuencia guía [61]. Posteriormente se introduce una nueva secuencia usando los mecanismos endógenos de reparación de ADN [54], como son el mecanismo de unión de extremos no homólogos (Non Homologous End-Joing) y el mecanismo de recombinación homólogo (Homologous Recombination) [61, 62].
La edición del genoma utilizando la tecnología CRISPR-Cas9 está siendo implementada para generar mutaciones específicas, y así simular enfermedades en células madre pluripotentes inducidas (iPSC) [63]; permitiendo estudiar diferentes tipos de células que simulan comportamientos fenotípicos de enfermedades como resultado del desequilibrio entre estados de enfermedad. Un ejemplo de esta implementación es el desarrollo de cardiomiocitos derivados de iPSC obtenidas a partir de pacientes que presentaron mutaciones específicas para enfermedades cardiacas [61]. Es posible que, en un futuro, este tipo de enfoque permita la obtención de sistemas celulares, que representen fenotípicamente algunas enfermedades, y así usar estos sistemas en imagenología de células vivas para la evaluación de compuestos.
Imagenología de células vivas para la monitorización de respuestas fenotípicas desencadenadas por interacción entre compuesto candidato y diana terapéutica
Una vez definidos los compuestos que serán evaluados, el sistema celular que será utilizado, la o las dianas con las cuales se espera interacción y los cambios fenotípicos relevantes para observar respuesta de interacción, se realiza el rastreo primario (primary screening). Este permite determinar el grado de interacción de los compuestos con la o las dianas terapéuticas.
Las técnicas de microscopía de fluorescencia de alto rendimiento en plataformas para imagenología de células vivas, ha facilitado la identificación, cuantificación y monitorización de procesos celulares [64-66]. Por lo cual, es una herramienta útil en el cribado de compuestos candidato. Ofrece alta resolución espacial y temporal de procesos celulares relevantes, siempre y cuando el sistema celular simule uno o varios fenotipos característicos, o aborde ciertos factores moleculares determinantes del desequilibrio de estados de enfermedad. Por ejemplo: Durante la infección del virus Dengue (DENV), los compartimentos celulares alterados pueden ser monitorizados y analizados individualmente (caracterizados por segmentación y cuantificación de imágenes) [67]. El compuesto evaluado dirigido a blancos celulares, en este caso denominado antiviral, debería impedir la progresión del patrón alterado o contribuir a la recuperación del fenotipo normal.
Para poder monitorear la respuesta de interacción, es necesario seleccionar un marcador fenotípico (sustancia cuantificable a partir de fenómenos físicos como la absorción o emisión de fotones a ciertas longitudes de ondas electromagnéticas). Las sondas y las etiquetas fluorescentes codificadas genéticamente son los más utilizados [68]. El máximo número de marcadores utilizados de manera simultánea, por sistema celular, depende de la capacidad técnica de los equipos de microscopía y del nivel de exposición según la duración de la formación de la imagen (esto evita fototoxicidad) [26, 68].
La utilización de sondas fluorescentes ha permitido, por ejemplo, el estudio de la dinámica del glutatión [69], que es decisiva en alteraciones celulares que conllevan al desarrollo de enfermedades, puesto que mantiene y modula la homeostasis redox [70]. Estas sondas, que son de respuesta rápida y reversible a glutatión intracelular, se basan en mecanismos de transferencia de energía por resonancia de fluorescencia (FRET) [69]. Este mecanismo consiste en la transferencia de energía, entre un marcador molecular donador inicialmente excitado y un marcador aceptor. La transferencia de energía que origina la señal a detectar depende de la distancia entre los marcadores y su orientación relativa en el espacio [71].
El uso de etiquetas fluorescentes codificadas genéticamente está dirigido al marcaje de proteínas decisivas en el fenotipo estudiado. Estas etiquetas son insertadas en la célula mediante un proceso de transfección (figura 3A). Esta estrategia de marcaje ha permitido observar, por ejemplo, la interacción coordinada y simultánea de diferentes orgánulos de sistemas celulares (como el aparato de Golgi, gotas lipídicas, lisosomas, mitocondrias, peroxisomas y retículo endoplásmico), en respuesta a diferentes fenómenos desencadenados por interacciones compuesto-diana (figura 3C) [72].
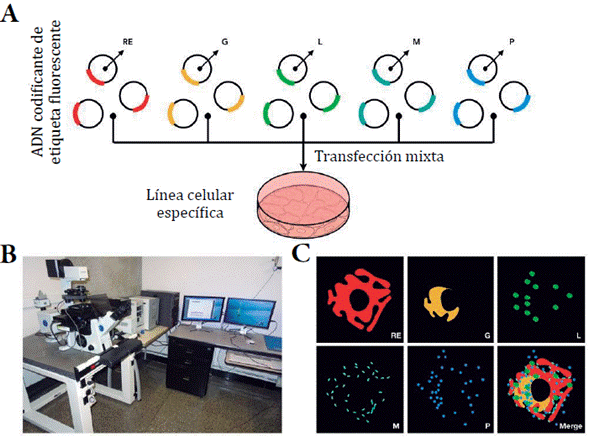
Figura 3 Proceso de transfección y monitorización del interactoma. A. Transfección mixta de ADN codificante de etiqueta fluorescente. Retículo endoplásmico (RE); golgi (G), lisosoma (L), mitocondria (M), peroxisomas (P). B. Microscopio confocal de disco giratorio. Permite la monito-rización de células vivas. c. imágenes y videos obtenidos como producto de la monitorización.
Durante la monitorización, cada compuesto evaluado tendrá una respuesta fenotípica distinta como reflejo del grado de interacción. Si lo que se evalúa es una colección de compuestos, es posible agrupar las respectivas respuestas en grupos de imágenes según el cambio fenotípico generado por cada compuesto [68]. Por ejemplo, es posible agrupar respuestas como la dinámica de distribución subcelular de orgánulos [67], nivel de expresión de proteína blanco según la intensidad fluorescencia [73], eventos apoptóticos [74] o comportamiento del interactoma entre orgánulos [72]. Posteriormente se transforman las imágenes en datos numéricos usando algoritmos de análisis cuantitativo, para luego transformar cada colección de datos en vectores de perfil fenotípico [26, 68]. Estos últimos son sometidos a su respectivo análisis estadístico para determinar la tasa de éxito en el cribado de los compuestos al afectar o modular el blanco terapéutico y dar paso al rastreo secundario (Secondary screening).
Un ejemplo útil de este proceso sería la monitorización de la distribución mitocondrial alrededor de los núcleos de células infectadas con DENV, generada por el efecto de antivirales candidatos. Es posible desarrollar dicho experimento gracias a la información obtenida en nuestro estudio cualitativo de la distribución mitocondrial en células vivas infectadas con DENV y un control con simulacro de infección [75]. Esta investigación ha logrado desarrollar una metodología automatizada para la cuantificación de la intensidad media de fluorescencia de una proteína mitocondrial en células con y sin infección [67]. Demostrando un comportamiento diferenciado entre ambos grupos de células (figura 4). Los videos de las mitocondrias tomados de las células vivas aportaron información cuantitativa, así como evidencia visual, con la cual se concluye que durante la infección con DENV la distribución mitocondrial es diferenciada respecto a células sin infección [75].
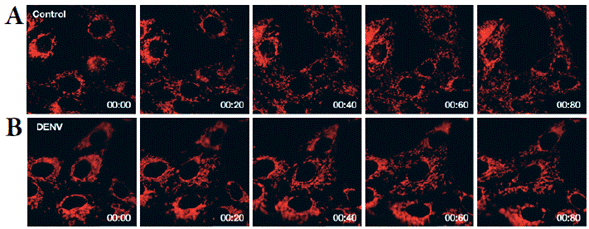
Figura 4 Distribución mitocondrial perinuclear diferencial en células infectadas con DENV y células sin infección. A. Dinámica mitocondrial de células sin infección (Mock). B. Dinámica mitocondrial de células infectadas con virus dengue (DENV).
Este tipo de investigaciones permitirán el cribado de compuestos antivirales con efectos en algunas funciones vitales de las células infectadas con DENV. Es ampliamente conocido que el DENV hace una parte de su ensamblaje en las membranas del retículo endoplásmico rugoso [76]. Resulta pues clave evaluar cómo potenciales antivirales alteran la distribución subcelular de este organelo y la dinámica de las vesículas, monitorizado en un curso temporal de células infectadas en comparación con células no infectadas. La capacidad de resolución de los equipos actuales permitirá tales avances investigativos.
CONCLUSIONES
El descubrimiento de fármacos implica varias subfases, incluida la evaluación de uno o más compuestos, durante la cual un sistema biológico simula un estado de pre-enfermedad reversible por modulación de dianas terapéuticas, y el rediseño estructural del compuesto, cuando se modifica la estructura para mejorar el grado de interacción con la diana. Sin embargo, la obtención de resultados prometedores está mediado por la estructura molecular del o de los compuestos a evaluar y por la representación adecuada de procesos patológicos a partir de dianas terapéuticas modulables en sistemas celulares. Las propiedades que determinan el grado de interacción compuesto-diana deben ser tenidas en cuenta antes de iniciar cada subfase del descubrimiento. La metodología experimental subyacente a esta fase implica un alto rendimiento para su monitorización, la cual puede ser proporcionada por la imagenología de células vivas. Después de la interacción compuesto-diana, la modulación puede resultar en el desencadenamiento de un fenotipo como respuesta. El cambio fenotípico del sistema celular, que media el equilibrio entre estados de enfermedad, proporciona los datos necesarios para realizar seguimiento por captura de imágenes. Por lo tanto, la monitorización por imagenología de células vivas se fundamenta en la adecuada selección de un fenotipo resultante a la modulación generada por el compuesto evaluado.
Aunque el descubrimiento de fármacos enfrenta diferentes retos relacionados con la caracterización de compuestos candidato, la identificación de dianas y con la monitorización de su interacción; cada uno de estos avanza en simultáneo al desarrollo de:
Tecnologías computacionales para el diseño estructural de compuestos.
Tecnologías para captura de imagen en células vivas.
Estrategias para la generación de sistemas celulares capaces de ofrecer información relevante para las enfermedades. Por lo tanto, es posible implementar estrategias, como la aquí propuesta, que integren el estudio racional de compuestos, la fisiopatología celular y la monitorización eficiente de la respuesta entre compuestos y dianas. Quizá solo así sea posible avanzar en la búsqueda nuevos tratamientos farmacológicos sin fracasar en ello.

