











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Coronaviruses are enveloped positive single-stranded large RNA viruses that infect humans, but also a wide range of animals [1]. According to their morphology as spherical virions with a central nucleus and surface projections that resemble a solar corona, they were called coronaviruses [2]. There are four subfamilies, namely alpha, beta, gamma, and delta coronavirus [1]. The size of the genome varies between 26 kb and 32 kb [3]. Among the coronavirus subtypes that can infect humans, beta-coronaviruses can cause serious illness and death, while alpha-coronaviruses cause asymptomatic or mildly symptomatic infections [1]. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the B-lineage of beta-coronaviruses and is closely related to the SARS-CoV virus [1]. There are four major structural proteins encoded by the coronaviral genome in the envelope, one of which is the spike protein (S) that binds to the angiotensin-converting enzyme 2 (ACE2) receptor and mediates subsequent fusion between the envelope and host cell membranes to aid viral entry into the host cell [2].
The world is currently facing a SARS-CoV-2 pandemic, declared by the World Health Organization (WHO) Emergency Committee on March 11, 2020, based on rising case notification rates in different countries. This virus has spread to 188 countries as of August 10 and has infected more than 19.995.057 people. The case detection rate changes daily and can be tracked in near real-time on the website provided by Johns Hopkins University and other forums [4]. Most worrying is that the disease has posed a serious threat to global public health, by requiring the development of safe and effective prophylactic and therapeutic drugs against the infection of its causative agent, SARS-CoV-2, also known as the new coronavirus 2019 (2019-nCoV) [3]. Therefore, the lack of specific medications to prevent and treat an attack is an important need at the present time, considering that the available treatment is clearly symptomatic [5]. Drug discovery can also be accelerated by applying new detection techniques, including computational approaches, to identify antivirals that could be ready for large-scale clinical trials in a matter of weeks [6].
The main pharmacological targets for coronavirus (CoV) are spike protein, envelope protein, membrane protein, protease, nucleocapsid protein, hemagglutinin esterase, and helicase, for which drug design can be considered. There are 16 other non-structural proteins (NSP) that can also be considered from the perspective of drug design including RNA-dependent RNA polymerase (RdRp), coronavirus main protease (3CLpro), and papain-like protease (PLpro). These proteins are important because upon entering host cells, the viral genome is released as a single-stranded positive RNA. Subsequently, it is translated into viral polyproteins using host cell protein translation machinery, which is then cleaved into effector proteins by viral proteins 3CLpro and PLpro. PLpro also behaves like a deubiquitinase that can de-position certain proteins from the host cell, including interferon factor 3 and NF-KB, resulting in immune suppression. RdRp synthesizes a full-length negative-strand RNA template to be used by RdRp to produce more viral genomic RNA [7]. 3CLpro is a key CoV enzyme, which plays a key role in mediating viral replication and transcription, making it an attractive drug target for this virus [7-8]. This protein was selected to be used as a pharmacological target in this study. Lopinavir is an antiretroviral drug used in patients with HIV and Remdesivir is an antiviral prodrug used to treat Ebola virus disease, currently these two drugs have been proposed as antivirals for SAR-CoV-2. Lopinavir and remdesivir are antiviral agents [7].
Therefore, the main structural proteins and NSP can play an important role in the perspective of drug design. However, the occurrence of frequent recombination events is an important deterrent towards the development of vaccines and medications specific to CoV [9]. The objective of this study is to use in silico approach to propose antiviral compounds potential for SARS-CoV-2 and SARS-CoV and drug-like properties predictions.
METHODS
Equipment and Software
Drug Likeness Tool (DruLiTo 1) was used to detect molecules with ADME properties. AutoDock Vina was the primary docking programs used in this work [10]. The preparation of the PDBQT files and determination of the grid box size was carried out using Auto-Dock Tools version 1.1. Chimera was utilized to prepare protein structures. PyMol (DeLano Scientific LLC, USA) was employed to visualize protein-ligand complex structures. The identification of protein residues that interact with small molecules was carried out employing LigandScout. Computational studies were carried out using a machine running on Inter Core i7 4.0GHZ, a processor with 8GB RAM and 1TB hard disk.
Selection of molecules with properties drug-likeness
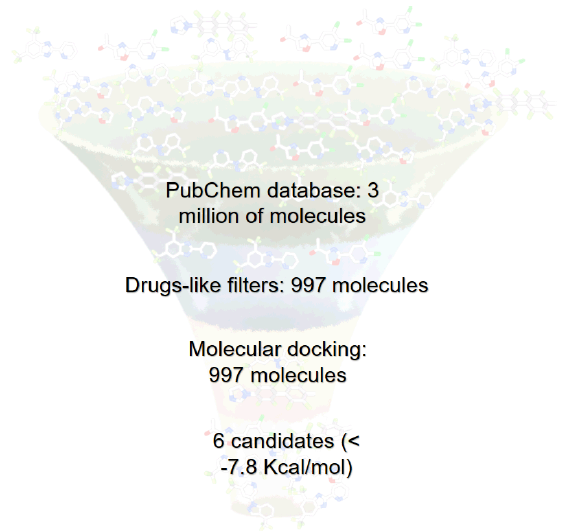
The database PubChem was used to download 3 million molecules deposited during 2015 and 2016. These chemicals were filtered considering those having characteristics of drugs-like, to filter molecules were used free software Drug Likeness Tool (DruLiTo 1). The rules Lipinski that predict if a compound is more likely to be membrane-permeable and easily absorbed by the body [11]. Veber rule, to predict good oral bioavailability [12]. Ghose filter that defines drug-likeness constraints [13]. The molecules that passed these filters were selected for molecular docking. The criteria that were taken into account were: molecular weight ≤ 500, Log P ≤ 5, H-Bond donor ≤ 5, H-bond acceptor < 10, rotatable bond ≤ 10, polar surface area ≤ 140, Atom-Count from 20 to 70 and refractivity from 40 to 130. Of the three million molecules discharged in PubChem, only 997 passed the drug-like filters, that is, they possess drug-like properties.
Protein structure preparation
The 3D structure of proteins: COVID-19 main protease and SARS coronavirus main peptidase were downloaded from Protein Data Bank (PDB: 6LU7 and 2GTB, respectively), both obtained by x-ray diffraction, and prepared with Chimera. The water molecules and other ligands present in the proteins were eliminated. Proteins were minimized using atomic partial charges by Kollman method, which describes the potential of the system in terms of the energy positions of the atoms and is parameterized for proteins and nucleic acids [14]. MGLTools 1.5.0 software was utilized to convert structures from PDB to PDBQT format, adding polar hydrogens and assigning Kollman partial charges [14].
Protein-ligand docking
Molecular docking was performed using AutoDock Vina is a virtual molecular docking and screening program. Vina uses a sophisticated gradient optimization method in his local optimization procedure. The gradient calculation effectively provides the optimization algorithm with a "sense of direction" from a single evaluation. By using multithreading, Vina can further speed up execution by leveraging multiple CPUs or CPU cores. The docking site for ligands on evaluated proteins was defined by establishing a cube with a sufficient dimension to cover the complete protein, with a grid point spacing of1 A for the proteins. Three runs were carried out by ligand, and for each run the best pose was saved. Finally, the average binding affinity for best poses was accepted as the binding affinity value for a particular complex [14]. The three-best protein-ligand complexes were selected, those that showed the highest affinity value.
Identifying interactions
Three ligand structures were selected and coupled with proteins to identify residues of proteins that interact with small molecules, using Ligand scout software. This program proposes the number and type of primary existing ligand-residue interactions on the protein active site. For this, the coordinates generated during molecular coupling are used, to identify the amino acids participating in the protein-ligand interaction.
RESULTS
In this study of the 3 million new molecules selected, only 997 met drug-like properties (rules Lipinski, Veber rule, and Ghose filter), which were used to make molecular docking. These molecules possess ADME properties and also have a high affinity for all proteins (molecules with scores below -7.8 kcal/mol) (table 1 and figure 1), this value represents the affinity of a protein for a ligand as a semi-quantitative parameter related to the inverse of the dissociation constant. Therefore, only the three molecules for each protein with higher affinity values were selected as possible candidates.
Table 1 Drug-like properties and AutoDock-calculated affinities obtained for docking of best molecules on proteins of COVID-19 (< -7.8 kcal/mol).
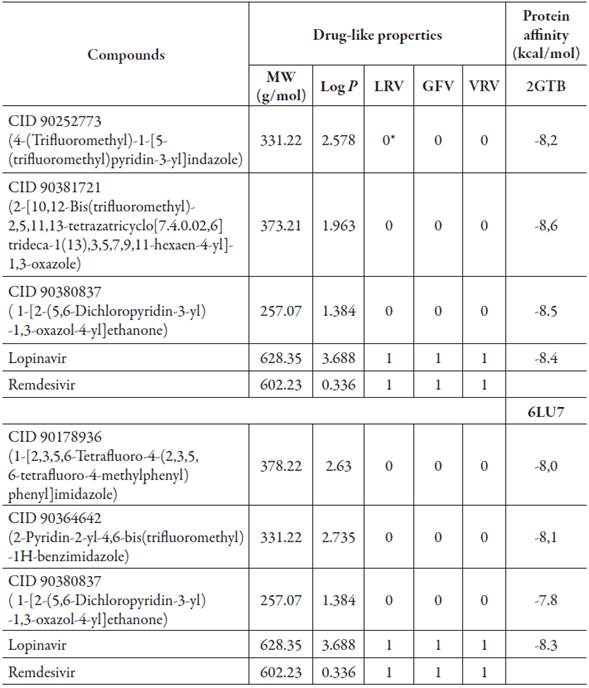
MW: Molecular weight
Log P: Logarithm of octanol/water partition coefficient
LRV: Lipinski rule of 5 violations: Maximum is 4 violations
GF: Ghose Filter violation
VR: Vebers Rule violation
*This value refers to the number of violations found for each drug-like filter evaluated.
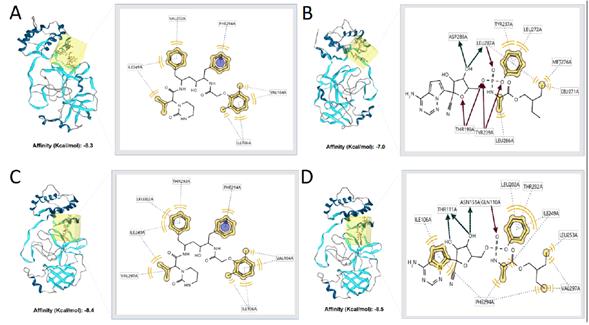
The three-best protein-ligand complexes were selected and are shown in figures 2 and 3 for each protein, SARS coronavirus main peptidase (2GTB), and COVID-19 main protease (6LU7). 6LU7 is the main protease (Mpro) found in SARS-CoV-2 and 2GTB is the main protease found in the SARS-CoV [15]. With both proteins, blind molecular docking was carried out, that is, docking over the entire surface of the protein. Furthermore, molecular docking was performed using the same protocol with two antiviral drugs, in order to validate the binding site of the ligands evaluated by comparison with lopinavir and remdesivir.
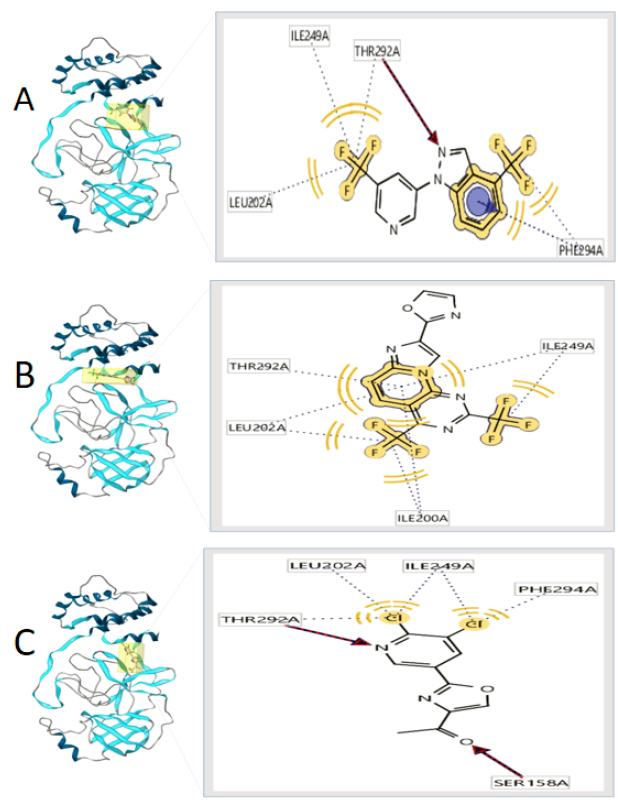
Source: Own elaboration.
Figure 2 3D view and interacting residues present in proteins 2GTB. A: 2GTB -90252773, B: 2GTB -90381721, C: 2GTB -90380837.
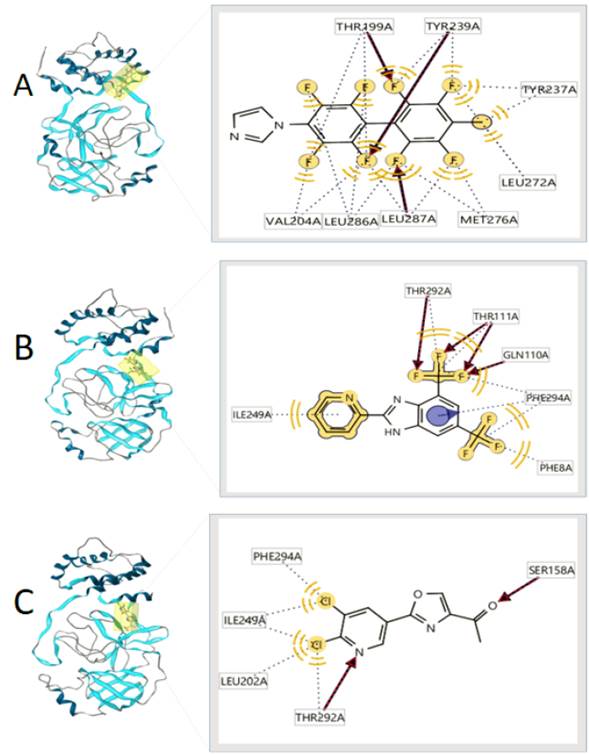
Source: Own elaboration.
Figure 3 3D view and interacting residues present in proteins 6LU7. A: 6LU7 -90178936, B: 6LU7 -90364642, C: 6LU7 -90380837.
Table 2 shows the amino acids found in the active site pockets of 6LU7 and 2GTB in the complex with two reported inhibitors (lopinavir and remdesivir) and the selected new molecules selected.
Table 2 Interacting residues 2GTB and 6LU7 protein - inhibitor complex in comparison with ligands studied.
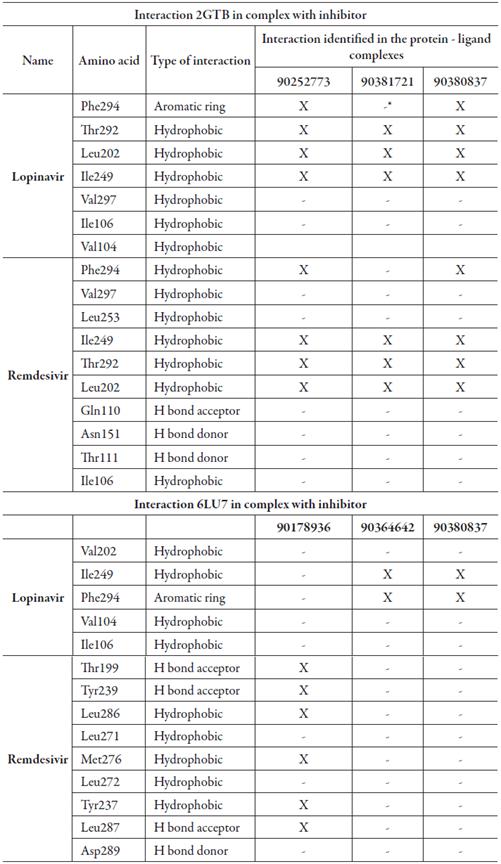
*The presence of the amino acid in the protein-ligand complex of the proposed candidates was not identified.
The affinity values and three-dimensional view of the complexes with lopinavir and remdesivir are shown in figure 4. Lopinavir and remdesivir are two antiviral drugs against a clinical isolate of SARS-CoV-2 in vitro (7). In this study, we have used them to check the ability of the proposed new molecules to bind to the same active site of the proteins evaluated to which these two antiviral agents used in the treatment of SARS-CoV-2 and SARS-CoV bind. Being able to identify that the amino acid residues that participate in the formation of the protein-ligand complex were also identified in the protein-ligand complexes of the molecules with the best affinity values (table 1 and 2). The predominant type of interaction was the hydrophobic type interaction.
DISCUSSION
SARS-CoV-2 represents a pandemic threat to public health because there is no treatment to counteract the virus [16]. So the search for new molecules with antiviral properties is imperative [7]. The present study focused on the main proteases in CoVs (3CLpro/Mpro), especially PDB ID 6LU7, as potential target proteins for SARS-CoV-2 treatment [15] and is the main peptidase of the SARS coronavirus (SARS-CoV M (pro)), which plays an essential role in the life cycle of the virus and is a main target for the development of anti-SARS agents [17]. Xu et al. [18] mentioned that the main protease in 2019-nCoV shares 96% similarity with that in SARS0 [14]. To make comparisons, these two proteins were used.
In the search for new molecules that can be used as antivirals, it is important to predict the similarity of the drugs, as well as the intestinal absorption capacity and bioavailability requirements [19]. In this study, the selected chemical compounds meet these. As for the affinities obtained, they are comparable with those obtained for lopinavir and remdesivir. Khaerunnisa et al., [15] reported that the binding energies obtained from the docking of 6LU7 with native ligand, nelfinavir, lopinavir, kaempferol, quercetin, luteolin-7-glucoside, demethoxycurcumin, naringenin, apigenin-7-glucoside, oleuropein, curcumin, catechin, epicatechin-gallate, zingerol, gingerol, and allicin were -8.37, -10.72, -9.41, -8.58, -8.47, -8.17, -7.99, -7.89, -7.83, -7.31, -7.05, -7.24, -6.67, -5.40, -5.38, and -4.03 kcal/mol, respectively. As can be seen, the affinity values reported using AutoDock and the same protein are comparable with those obtained in this study.
Several authors [15, 20, 21] have reported new molecules with antiviral properties against SARS-CoV-2 through molecular docking with affinity values comparable to those obtained in this study. For the main peptidase of the SARS coronavirus, the protein-ligand complex (2GTB -90252773, B. 2GTB -90381721, C. 2GTB -90380837) show that all bind to the same active site including lopinavir and remdesivir, involving the same amino acids and the same type of interactions, being the interactions of hydrophobic type the main ones found. The protein complexes binding to the 6LU7 protein show that the 6LU7-90364642 and 6LU7-90380837 complexes bind to the same site as lopinavir, but the 6LU7-90178936 complex interacts at a different site together with remdenavir, which leads us to think of a possible allosteric site of interaction, in which other amino acids with predominantly hydrophobic interactions are involved. Chauhan [21], reported that remdesivir couples with 6LU7 with an affinity value of -13.1. Khalifa et al. [22], reported that tellimagradin interacts within the protease binding pocket with the amino acids Phe294, Thr292, Ile249, among others, which were also identified in our protein-ligand complexes. These results suggest that small molecules identified by in silico approaches and with drug-like properties may be useful candidates for the development of new antiviral drugs. These molecules are available in the PubChem database and can be used to carry out experimental studies.
The results obtained are of great importance because they allow us to identify potential molecules with antiviral characteristics using an in silico approach, which reduces the time and budget required in the laborious search for a treatment to stop the progression of this disease that is affecting millions of people in the world. In this study, it is recommended to continue to a next phase in drug development, which consists of conducting in vitro and in vivo evaluation with the proposed candidates, to develop new drugs against SARS-CoV-2 and SARS-CoV.
CONCLUSIONS
The protein-ligand complexes 2GTB -90252773, 2GTB -90381721, 2GTB -90380837, 6LU7-90364642, 6LU7-90380837 and 6LU7-90178936 were those that showed the highest affinity values (< -7.8), with the ability to interact in the same binding sites for lopinavir and remdenavir, being potential candidates with drug-like properties to act as antivirals against SARS-COV and SARS-CoV-2. These molecules can be evaluated by pharmacological experimentation.

