











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCCIÓN
Uno de los grandes retos de la industria farmacéutica es la administración de principios activos que presentan características biofarmacéuticas que limitan su absorción oral directa mediante formas farmacéuticas tradicionales [1]. Se han descrito muchas estrategias para aumentar la velocidad de disolución de los fármacos mediante la reducción de su tamaño de partícula y la formación de sales, utilizando tensoactivos, ciclodextrinas, liposomas o nanopartículas [2, 3]. Un enfoque relativamente nuevo para los fármacos poco solubles son las formulaciones basadas en lípidos, particularmente, los sistemas autoemulsionables de liberación de fármacos [4]. Los sistemas autoemulsionables de liberación de fármacos (SEDDS por sus siglas en inglés) son sistemas transparentes, isotrópicos y, generalmente, están constituidos de fármaco, vehículo oleoso, tensoactivo y cotensoactivo. Esta tecnología se ha empleado para aumentar la solubilidad y esto causa la biodisponibilidad de muchos fármacos poco solubles en agua como la filantina, celastrol, ketoprofeno e hidrocortisona [4]. Los sistemas lipídicos se han convertido en un área de oportunidad para mejorar la solubilidad de los compuestos lipofílicos, ya que se ha observado que son capaces de incrementar la solubilidad gastrointestinal. La liberación de fármacos desde un SEDDS por vía oral ocurre por medio de una dilución en los fluidos del tracto gastrointestinal (TGI) e interactúa con las enzimas y los materiales endógenos como las sales biliares y la lipasa pancreática [5, 6], por ende, son sometidas a varios procesos de digestión cuyo resultado es la formación de las diferentes estructuras coloidales, tales como las vesículas lipídicas y las micelas mixtas. Esta transformación estructural es crucial para la solubilización de los fármacos y, por lo tanto, para evitar la precipitación del principio activo, proporcionando un entorno propicio para mejorar la absorción del fármaco [7].
Las formulaciones lipídicas se clasifican en cuatro grupos, donde las formulaciones tipo III (A y B) son los sistemas que contienen tensoactivos solubles en agua y cotensoac-tivos en proporciones mayores que la fase lipídica, mezclas que corresponden a sistemas autoemulsionables, mientras que los sistemas tipo IV corresponden a mezclas de tensoactivos y cotensoactivos solubles en agua que forman estructuras micelares [8]. La gran mayoría de estas formulaciones lipídicas son líquidas ya que los excipientes utilizados son líquidos o semisólidos [9], por lo tanto, estas formulaciones se incorporan en cápsulas de gelatina blanda o dura para obtener una forma farmacéutica sólida. Sin embargo, existen diferentes técnicas como la adsorción sobre un acarreador sólido o procesos de fusión (granulación o extrusión), para convertir estas formulaciones líquidas en formas de dosificación sólidas [10].
Los componentes de los SEDDS pueden ser de origen natural o sintético. De hecho, la combinación de las características de los ácidos grasos naturales con las de muchos lípidos sintéticos podría mejorar el funcionamiento de las matrices lipídicas diseñadas a la medida, con el objetivo de controlar sus propiedades de liberación [11, 12], similares a aquellas observadas en las microemulsiones [13], tras una dilución en un medio acuoso o fluidos gastrointestinales [14, 15] seguidas de una agitación suave proporcionada por la motilidad digestiva del estómago y del intestino [16]. Estos sistemas son eficaces siempre que estén diseñados con una selección cuidadosa de los excipientes, por su relación con las propiedades del fármaco [17], que generalmente es la necesidad de disolver la dosis requerida en una forma de dosificación unitaria para promover una dispersión rápida, una autoemulsificación de la formulación durante el paso a través del tracto gastrointestinal y la capacidad para mantener una solubilización efectiva del fármaco durante la dispersión y la digestión de la formulación [18]. Los sistemas deben su capacidad de carga (solubilidad del principio activo), así como su estabilidad, no sólo a las propiedades fisicoquímicas de los componentes individuales, sino también a los factores que resultan de la formación del sistema autoensamblado en partículas coloidales, tales como el tamaño de partícula, potencial zeta y viscosidad [19]. Su capacidad de formar emulsiones con glóbulos de tamaño nanométrico le confieren un área superficial grande para favorecer la absorción del fármaco [20, 21].
La indometacina es un potente fármaco Aine lipófilo derivado del ácido indolacético, que se prescribe para el tratamiento de: osteoartritis, artritis reumatoide, espondilitis anquilosante, dolor agudo de hombro, como tratamiento de la artritis gotosa aguda, de la artropatía degenerativa de la cadera, dolor lumbosacro, así como en el manejo de la inflamación, dolor e hinchazón posoperatorios ortopédicos, maniobras de reducción e inmovilizaciones, actualmente, se utilizan diferentes vías de administración para el tratamiento de estas enfermedades, como la oral, tópica y la inyectable [14, 22]. La indometacina tiene baja solubilidad acuosa, de acuerdo con el sistema de clasificación biofarmacéutica (SCB) es un fármaco de clase II. Este fármaco posee una reacción adversa irritante en el TGI, debido a su baja solubilidad y prolongado contacto con la mucosa gástrica, lo cual está relacionado con la dosis y el tiempo de tratamiento [14, 22, 23].
El uso de formulaciones basadas en lípidos en la administración oral de fármacos antiinflamatorios no esteroideos (Aines) como la indometacina (que a su vez son gastroirritantes) tiene la ventaja potencial de que el fármaco no sólo podría permanecer en disolución durante todo el transcurso en el tracto gastrointestinal (TGI), sino que los lípidos podrían actuar también como una cubierta protectora de la mucosa y los tensoactivos aumentan la permeabilidad del lumen intestinal [18].
La indometacina se encuentra en el mercado en diferentes presentaciones, en cápsulas de 25 mg, cápsulas de liberación prolongada de 50 o 60 mg, supositorios de 100 mg y otras presentaciones como crema y en aerosol. La dosis recomendada es de 25 a 50 mg de 2 a 3 veces al día [24].
En este trabajo hemos propuesto la incorporación de indometacina en sistemas auto-emulsionables de liberación de fármacos, los cuales tienen la finalidad aumentar la solubilidad y velocidad de disolución en un medio acuoso, así como demostrar el incremento en la difusión a través de las membranas de diálisis. Las características propias del sistema pueden conferir ventajas a la formulación tipo SEDDS comparada con los productos comerciales de indometacina.
METODOLOGÍA
Fármaco y excipientes
Indometacina, propilenglicol y polietilenglicol se compraron a la Distribuidora Química Lufra, S.A. Labrasol (macrogol-8-glicérido de caprilocaproilo), Labrafil M2125CS (macrogol-6-glicérido de linoleoilo), Gelucire 48/16 (estearato de macro-gol), Lauroglicol 90 (monolaureato de propilenglicol [tipo II]), Lauroglicol FCC (monolaureato de propilenglicol [tipo I]), Maisine CC (monolinoleato de glicerilo), Labrafac lipophile WL1349 (triglicéridos de cadena media) y Transcutol HP (monoetil éter de dietilenglicol), todos de Gattefossé (Francia), fueron proporcionados en donación por Químicos Lyontec, S.A. Kolliphor EL (aceite de ricino 35-polioxilo) fue donado por el distribuidor en México de BASF (Alemania). Tween 80 (polisorbato 80) fue provisto por Merck México, S.A. de C. V.
Método de cuantificación para indometacina
Una disolución de indometacina fue preparada a una concentración de 30 μg/mL y se realizó un barrido de absorbencia de 200 a 400 nm para determinar la longitud de onda de máxima absorción. Una vez seleccionada la longitud de onda de trabajo se elaboró una curva de calibración realizando una prueba de linealidad y precisión del sistema.
La linealidad del sistema se evaluó preparando por triplicado una curva de calibración de 5 niveles, por medio de diluciones sucesivas a partir de una disolución de 30 μg/mL. La precisión del sistema se determinó midiendo la respuesta analítica de 6 diluciones individuales a una misma concentración a partir de una disolución de 30 μg/mL. Se evaluó la especificidad del método analítico realizando barridos de absorbencia para cada excipiente utilizado.
Solubilidad de la indometacina en los excipientes
A una cantidad conocida de cada excipiente se añadió un exceso de indometacina, las mezclas se agitaron utilizando un agitador tipo vórtex (Lab-line Instruments) hasta obtener una mezcla turbia. Las mezclas se colocaron en un baño de agua ( Julabo modelo SW22) con agitación a 175 rpm, a 25 °C, durante 72 h para solubilizar la mayor cantidad de indometacina y alcanzar el equilibrio de saturación. La cantidad de indometacina solubilizada a 72 h se cuantificó utilizando un espectrofotómetro UV-Vis (Shimadzu, modelo UV-1201).
Preparación y caracterización de mezclas lipídicas
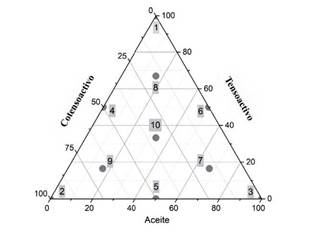
Por medio de un diagrama de fases ternario (figura 1) se determinaron las cantidades de cada uno de los componentes de la mezcla y se ubicaron 10 puntos que cubran la mayor cantidad de superficie del diagrama. A cada mezcla lipídica se le adicionó la dosis de indometacina (25 mg/mL) y se agitó hasta mezclar perfectamente todos los componentes, los sistemas se mantuvieron en agitación constante de 175 rpm a 25 °C durante 48 h. Al terminar el tiempo se realizaron observaciones macroscópicas de los sistemas, tales como la miscibilidad entre los componentes de la mezcla y su capacidad para formar una emulsión en un medio acuoso, para esto se tomó una alícuota de 100 μL de la mezcla lipídica y se adicionó a 100 mL de agua destilada a 37 °C y con agitación de 100 rpm en una parrilla con agitación magnética (Cole-Parmer, modelo StableTemp).
Preparación e inspección visual de sistemas autoemulsionables
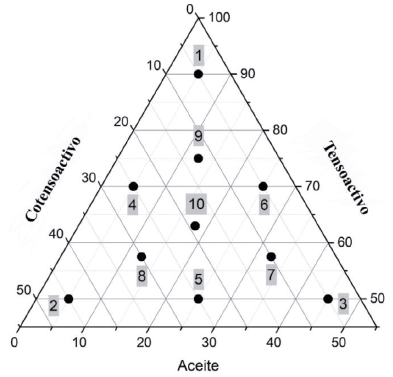
Una vez definida el área donde las mezclas lipídicas presentaron una capacidad de formar emulsiones en un medio acuoso con agitación suave, se eligió una nueva área de trabajo y se prepararon 5 series de sistemas cargados con la dosis de indometacina (25 mg/mL), utilizando el mismo tensoactivo y cotensoactivo y 5 vehículos oleosos diferentes uno para cada serie de 10 mezclas distintas localizadas en un diagrama ternario (figura 2), los vehículos oleosos utilizados fueron Gelucire 48/16, Tween 80, Kolliphor EL, Lauroglicol 90 y Labrafil M2125CS. Una vez preparados los sistemas, se verificó que fueran traslúcidos y se excluyeron aquellos que a temperatura ambiente estuvieran sólidos o que no hubieran solubilizado la dosis de indometacina.
Eficiencia de la autoemulsificación
Para la prueba de eficiencia de autoemulsificación se tomó una alícuota de 500 μL de la formulación y se agregó a un volumen de 100 mL de agua destilada a 37 °C contenidos en un matraz Erlenmeyer con agitación de 100 rpm; se registró el tiempo que tarda la formulación en dispersarse por completo en dicho volumen. Para cada sistema el procedimiento se realizó por triplicado.
Estabilidad física
Los sistemas fueron sometidos a tres pruebas para evaluar su estabilidad, la prueba de centrifugación se realizó en una centrífuga (Eppendorf, modelo 5427) a 5000 rpm durante 30 minutos, al término del tiempo se verificó si las formulaciones presentaron alguna inestabilidad, como separación de fases o precipitación del fármaco. Las formulaciones que no mostraron inestabilidad fueron sometidas a un ciclado de temperatura de enfriamiento-calentamiento, la cual consiste en 6 ciclos de 48 h de 4 °C a 40 °C. Las formulaciones estables a estas temperaturas fueron elegidas para la prueba de estrés de congelación-descongelación [25], esta prueba de ciclado consistió en 3 ciclos de 48 h de -21 °C a 25 °C. Las formulaciones que fueron estables durante las pruebas de estabilidad física continuaron con la etapa de caracterización.
Distribución de tamaño de partícula, índice de polidispersidad y potencial zeta
La distribución del tamaño de la partícula, índice de polidispersión y potencial zeta de las formulaciones se determinó utilizando el equipo Zetasizer nano ZSP. El tamaño de partícula y el índice de polidispersión se determinaron en celdas desechables de plástico, la dispersión de la luz se controla a 25 °C en un ángulo fijo de 173°. Las mediciones del potencial zeta se realizaron en una celda capilar curva a 25 °C. Todas las determinaciones se realizaron por triplicado.
Perfiles de disolución y difusión en membranas de diálisis
Las formulaciones que presentaron los mejores resultados de tamaño de partícula, índice de polidispersidad y potencial zeta se eligieron para la prueba de perfil de disolución y difusión en membranas de diálisis (12 000 Da). Se utilizó medio de disolución de fosfatos a un pH 7,2; a una temperatura de 37,5 °C para ambas pruebas. La evaluación de los perfiles de disolución se realizó utilizando un disolutor (VanKel, modelo 10-1200) adecuado con el aparato I, con 750 mL de medio de disolución con una agitación de 100 rpm. Se aplicó el mismo método para el producto comercial y las formulaciones SEDDS, para estas últimas se colocó 1 mL de formulación SEDDS en las cápsulas de gelatina dura con la dosis de 25 mg de indometacina disuelta y se realizaron 7 muestreos de 5 mL sin reposición de medio de disolución durante 60 minutos. La evaluación de la difusión en la membrana de diálisis se realizó utilizando el aparato II con 750 mL de medio de disolución con una agitación de 75 rpm. Se utilizaron membranas de celulosa con 5 mL de volumen interno de cada sistema y se realizaron 11 muestreos sin reposición de medio en un tiempo total de 4 h. Para cada sistema de estudio se aplicó el mismo método por triplicado y las muestras fueron cuantificadas a 320 nm en un espectrofotómetro UV-Vis.
RESULTADOS
Método de cuantificación para indometacina
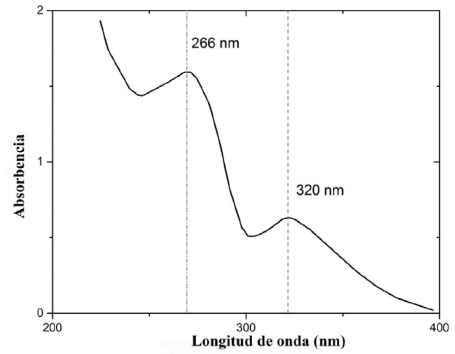
El barrido de absorbencia de la disolución de indometacina a una concentración de 31,56 μg/mL mostró dos máximos, correspondientes a 266 nm y 320 nm (figura 3). Este resultado puede analizarse como prueba de identificación de la indometacina ya que muestra un espectro parecido al reportado por Moffat; donde se observa un máximo de absorción a 318 nm en un medio de metanol acidificado [26]. El pico a 320 nm fue seleccionado para realizar las cuantificaciones correspondientes durante el desarrollo y evaluación de los sistemas de estudio, esto para tener la máxima sensibilidad y minimizar los errores, ya que la concentración a la que se preparó la disolución de indometacina genera valores de absorbencia alrededor de 0,6; mientras que en la longitud de onda de 266 nm se obtiene una absorbencia de 1,6; esto corresponde a una imitación de la técnica, ya que en disoluciones concentradas, las moléculas de soluto interaccionan entre sí debido a su proximidad y sus propiedades tales como la absortividad molar pueden cambiar [27].
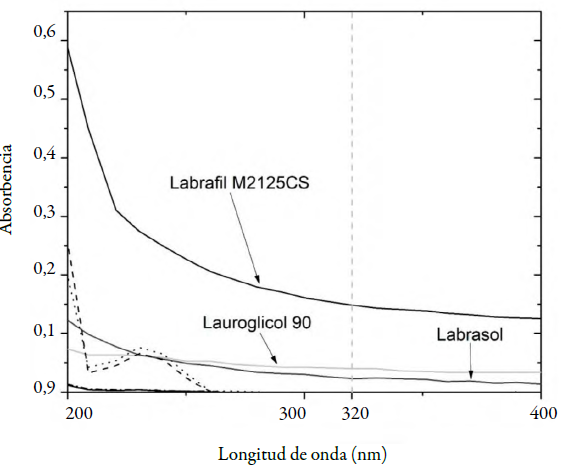
Se preparó por triplicado una curva patrón de indometacina con la finalidad de evaluar la linealidad del sistema, los resultados mostraron que el sistema se comporta de manera lineal en un intervalo de 5 a 40 μg/mL, el coeficiente de determinación obtenido fue 0,999 y el coeficiente de variación, menor a 2% para cada concentración de la curva. La precisión del sistema se evaluó realizando la cuantificación de 6 diluciones independientes a una concentración de 31,8 μg/mL, donde el coeficiente de variación entre las 6 mediciones fue menor a 1%, estos parámetros brindan la certeza de que el método analítico es eficiente para realizar la cuantificación del fármaco [28]. Los barridos de absorbencia correspondientes a los excipientes lipídicos mostraron que la mayoría de estos excipientes no absorben radiación en el mismo intervalo que la indometacina. Sin embargo, Labrasol, Lauroglicol 90 y Labrafil M2125CS mostraron absorción mínima insignificante respecto a la absorción de la indometacina que se adiciona en los sistemas lipídicos (figura 4).
Solubilidad de la indometacina en excipientes
Para evaluar la solubilidad de la indometacina se debe identificar y seleccionar una fase oleosa, tensoactivo y cotensoactivo con una buena capacidad de solubilización de la indometacina a temperatura de 25 °C para obtener el desarrollo óptimo de una formulación tipo SEDDS [29-31].
En la figura 5 se representa la solubilidad al equilibrio de la indometacina en los diferentes excipientes, se puede observar que Labrafac lipophile WL1349, Propilenglicol, Lauroglicol FCC y Maisine CC presentan una baja o nula capacidad para solubilizar a la indometacina, se evaluaron tensoactivos con valor bajo de HLB (Labrafil M2125CS y Lauroglicol 90) y valor alto de HLB (Labrasol, Tween 80 y Kolliphor EL), donde se puede observar que los tensoactivos de bajo HLB presentan un menor poder de solubilización de indometacina comparados con los tensoactivos de alto valor de HLB. Para el caso de los cotensoactivos como Transcutol HP y Polietilenglicol se puede observar que el primero de estos es el excipiente que presenta la mayor capacidad para solubilizar el fármaco. La evaluación de la solubilidad del fármaco en los diferentes excipientes es un parámetro de mucha importancia en el diseño de los sistemas tipo SEDDS estables, pues de esta manera se evitan problemas de precipitación durante su vida de anaquel y después de su dilución en la fase acuosa [4].
El Transcutol HP se seleccionó como cotensoactivo por su alta capacidad para solubilizar la indometacina. Al respecto, se reportó que puede ayudar a disolver grandes cantidades de tensoactivos hidrofílicos y de fármacos hidrofóbicos formulados en sistemas a base de lípidos, lo que representa una función importante en los SEDDS [25]. Este excipiente tiene el efecto de reducir aún más la tensión interfacial, al mismo tiempo que incrementa la fluidez de la interfase.
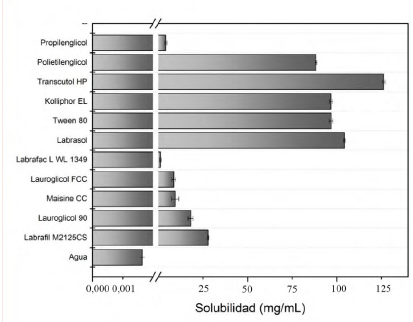
Por lo tanto, el resultado es el incremento en la espontaneidad para formar una emulsión [32]. El Labrasol fue seleccionado como el agente tensoactivo porque presentó la mayor solubilización de la indometacina, esta capacidad de solubilización de fármacos poco solubles en agua puede evitar la precipitación del fármaco dentro del cuerpo y promover que permanezca en disolución durante mayor tiempo durante en su paso por el TGI [25]. El Labrasol es un polioxil glicérido que está compuesto de una mezcla de mono, di y triésteres de glicerol y mono y diésteres de polietilenglicol (PEG). La capacidad disolvente del Labrasol está en función de la concentración de los grupos éster que tenga la molécula y las fracciones de PEG libres, así como por la longitud de la cadena hidrocarbonada, pues se reportó que las cadenas de triglicéridos saturadas, con 8 a 10 carbonos, presentan ventajas tecnológicas y fisiológicas [25], por lo que son una buena opción para usarlas en los SEDDS [32]. Los tensoactivos no iónicos como Labrasol, Gelucire y Kolliphor E son utilizados en el desarrollo de los SEDDS por contar con un control regulatorio y, por lo tanto, pueden ser utilizados como transportadores de fármacos para las diferentes vías de administración, tales como la oral, parenteral y la dérmica [11]. Estos excipientes también se caracterizan por tener valores menores de concentración micelar crítica (CMC), es decir, que a bajas concentraciones, estos ten-soactivos comienzan a formar agregados o estructuras coloidales, lo que favorece el proceso de la autoemulsificación.
El Gelucire es un estearato de polioxilo, con la misma mezcla de ésteres de glicerol y PEG que el Labrasol y Labrafil, pero sus cadenas hidrocarbonadas de ácidos grasos son de 18 carbonos y están saturadas, por lo cual su capacidad disolvente se ve ligeramente disminuida en comparación con los otros 2 polioxilglicéridos. Otros tensoactivos que solubilizaron en gran medida a la indometacina fueron el Kolliphor EL (aceite de ricino) y el Tween 80 (ésteres de ácidos grasos de sorbitán). Ambos son tensoactivos solubles en agua y de valores altos de HLB. Su capacidad disolvente está relacionada con su grado de etoxilación, mientras más polioxietilada se encuentre la molécula, mayor será la capacidad de disolver fármacos poco solubles en agua. Ambos excipientes fueron seleccionados como vehículos oleosos, ya que se pueden generar formulaciones de tipo IV, según la clasificación de Pouton, que indica que la mezcla de componentes hidrófilos: tensoactivos solubles en agua y codisolventes hidrofílicos, forman soluciones micelares tras la dilución en un medio acuoso [17] y con partículas de 20 nm o inferiores [33].
El Labrafil M2125CS también es un polioxilglicérido, con mezclas de mono, di y triésteres de glicerol y mono y diésteres de PEG y con un HLB menor en comparación con Labrasol [34], por tal razón fue seleccionado como vehículo oleoso. El Lauroglicol 90 es un tensoactivo insoluble en agua, su uso está reportado para los sistemas autoemul-sionables de tipo SEDDS y SMEDDS [35].
Preparación y caracterización de los sistemas lipídicos
En el diagrama de fases pseudoternario de la figura 1 se ubicaron 10 distintos puntos para cubrir la mayor área de trabajo y así poder tener excipientes puros, mezclas binarias y mezclas ternarias, donde cada punto indica la fracción en masa de cada componente. Los sistemas 1, 2 y 3 que se encuentran en los vértices son los excipientes puros, los sistemas 4, 5 y 6 son las mezclas binarias y los sistemas 7, 8, 9 y 10 son las mezclas ternarias.
Debido a que los excipientes utilizados para preparar las distintas formulaciones son de carácter oleoso, ningún sistema mostró inmiscibilidad de los excipientes dentro de la misma; sin embargo, en algunos sistemas no se logró solubilizar la dosis, motivo por el cual fueron descartados. A los sistemas restantes se les realizó la prueba de autoemulsificación con el propósito de establecer una zona de mayor probabilidad donde las mezclas tengan propiedades de realizar un proceso de dispersión espontánea característica de los sistemas SEDDS. Estos sistemas, al ser diluidos en medios acuosos, pueden tomar una coloración azulada, blanca-azulada o blanca-grisácea. Esta etapa de caracterización permitió establecer una zona delimitada por tener concentraciones de tensoactivo del 50-90%, aceite del 5-45% y cotensoactivo del 5-45%, las cuales se muestran delimitadas en la figura 2. La concentración de tensoactivo en las formulaciones autoemulsionables requeridas para formar y mantener un estado de emulsión en el TGI, según Kang et al. debe ser superior a 50% m/m de la formulación, pero depende de la naturaleza de los demás excipientes y del fármaco que se requiera solubilizar, por lo que estos porcentajes podrían cambiar [21]. El efecto de solubilización de la microemulsión debe atribuirse al uso de altas concentraciones de tensoactivo, por lo que se han estudiado las interacciones intermoleculares entre tensoactivos y fármacos para explicar los mecanismos de solubilización [36]. La presencia de cotensoactivos aumenta la fluidez de la interfase permitiendo que se puedan formar diferentes estructuras coloidales requeridas para formar la microemulsión [32].
Preparación e inspección visual de sistemas autoemulsionables
Una vez establecida la zona de autoemulsificación dentro del diagrama ternario (figura 2), se prepararon 5 series (A, B, C, D, E) de 10 sistemas cada uno, con una dosis de indometacina de 25 mg/mL. Todas las series presentaron en común el uso de Labrasol como tensoactivo y Transcutol HP como cotensoactivo, variando el vehículo para cada serie, la identificación de los sistemas se realizó asignando un número a cada vehículo complementario; A-Gelucire 48/16, B-Tween 80, C-Kolliphor EL, D-Lauroglicol 90 y E -Labrafil M2125CS (tabla 1).
Tabla 1 Composición de las formulaciones SEDDS cargadas con indometacina.
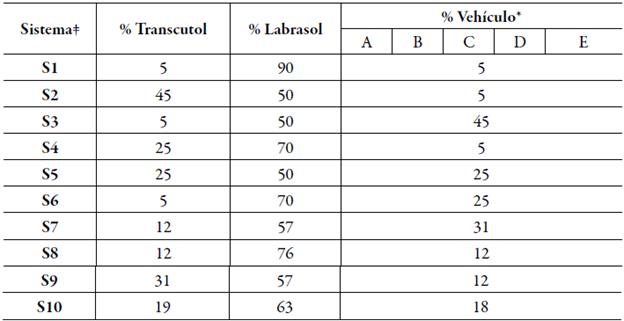
* A: Gelucire 48/16, B: Tween 80, C: Kolliphor EL, D: Lauroglicol 90 y E: Labrafil M2125CS, t S: A, B, C, D o E.
La inspección visual de los sistemas preparados es la primera prueba a la que son sometidas las formulaciones, pues algunas veces a simple vista se pueden observar inestabilidades o incompatibilidades entre los excipientes o entre los excipientes y el fármaco.
En la tabla 2 se encuentran los resultados de la inspección visual. Los sistemas que lograron solubilizar la dosis de indometacina sin presentar fenómenos de inestabilidad fueron seleccionados para las siguientes pruebas de caracterización, mientras que los sistemas que no cumplieron con lo anterior fueron excluidos. Como se puede observar en la tabla 2, la preparación de los sistemas A3, A5, A6, A7, A8 y A10 dio como resultado sistemas solidificados, en los que se pudieron observar partículas de Gelucire 48/16, pues este excipiente es sólido a temperatura ambiente. Esto se debe a la cantidad de Transcutol HP que es menor o igual a la cantidad de Gelucire 48/16, por lo tanto, el cotensoactivo en esta serie de sistemas es muy importante para mantener el sistema en estado líquido. El sistema D3 presentó un fenómeno de precipitación del fármaco ya que las cantidades de Transcutol HP y Labrasol no fueron las suficientes para solubilizar la dosis, por lo que estos sistemas fueron excluidos. Los sistemas A1, A4 y A9 presentaron una apariencia ligeramente opaca, mientras que todos los sistemas restantes presentaron una apariencia transparente.
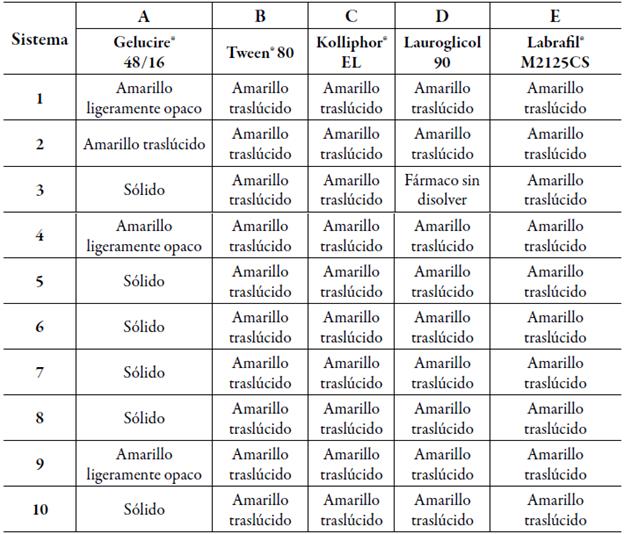
Eficiencia de la autoemulsificación y estabilidad física de los sistemas
Esta prueba tiene la finalidad de observar si las formulaciones lipídicas tienen la capacidad para formar una emulsión al entrar en contacto con un medio acuoso, únicamente con una agitación suave que simula los movimientos peristálticos, suficiente para la formación de los glóbulos in situ después de su administración oral [5, 6]. En la tabla 3 se registraron los tiempos de dispersión de los diferentes sistemas en medio acuoso, obteniéndose tiempos de 10 a 30 segundos, formando emulsiones ligeramente azuladas que son indicativas del proceso eficiente de formación de los sistemas nanométricos [25]. Los sistemas D5, D6, D7, E3, E5 y E7 fueron excluidos debido a que tardaron más de 2 minutos en dispersarse, además, al detener la agitación se pudo observar la formación de las gotas de aceite en la superficie.
Kang et al. reportaron sistemas SEDDS con los excipientes, Labrasol, Labrafil 1944 y Transcutol HP, como tensoactivo, vehículo oleoso y cotensoactivo, respectivamente; habiendo obtenido como resultado una zona de autoemulsificación similar a la generada por los sistemas propuestos en este trabajo [21]. Dichos sistemas presentaron una buena estabilidad física, así como excelentes características propias de los SEDDS, tales como tamaño de partícula no mayor a 2 Lim y una gran capacidad de carga del fármaco.
Tabla 3 Tiempo de autoemulsificación (s) de los sistemas cargados con indometacina.
| Sistema | Gelucire* 48/16 | Tween* 80 | Kolliphor* EL | Lauroglicol* 90 | Labrafil* M2125CS |
|---|---|---|---|---|---|
| 1 | 15,76 ± 2,14 | 15,40 ± 1,02 | 13,73 ± 2,11 | 18,97 ± 1,62 | 14,36 ± 2,20 |
| 2 | 10,53 ± 1,15 | 11,12 ± 0,28 | 9,16 ± 0,49 | 12,37 ± 1,11 | 12,09 ± 0,89 |
| 3 | ND | 14,52 ± 0,72 | 17,81 ± 2,94 | ND | ND |
| 4 | 10,24 ± 0,97 | 9,93 ± 0,23 | 9,84 ± 0,69 | 12,34 ± 3,59 | 13,26 ± 2,33 |
| 5 | ND | 12,41 ± 1,13 | 9,73 ± 1,41 | > 120 | ND |
| 6 | ND | 13,57 ± 1,11 | 12,30 ± 1,78 | > 120 | 17,96 ± 1,82 |
| 7 | ND | 12,75 ± 0,73 | 15,10 ± 2,52 | > 120 | ND |
| 8 | ND | 10,44 ± 0,68 | 12,91 ± 1,06 | 12,28 ± 0,87 | 17,52 ± 0,71 |
| 9 | 12,43 ± 0,54 | 10,16 ± 2,03 | 9,86 ± 0,35 | 16,07 ± 1,04 | 11,61 ± 0,99 |
| 10 | ND | 10,79 ± 1,69 | 11,25 ± 0,52 | 28,58 ± 2,76 | 18,05 ± 1,16 |
ND: no determinado. Cada valor representa la media ± D.E. (n=3).
Kallakunta et al. reportaron una serie de sistemas lipídicos que contienen Labrasol, Transcutol P y Gelucire 44/14, como tensoactivo, cotensoactivo y vehículo oleoso, respectivamente [37]. Los excipientes y sus proporciones son similares a los sistemas propuestos en este trabajo y en las pruebas de caracterización de las emulsiones se obtuvo como resultado tamaños de partícula de aproximadamente 100 a 200 nm, con valores menores a 0,3 del índice de polidispersión.
Los resultados de las pruebas de estrés a las que se sometieron los sistemas se muestran en la tabla 4. Prácticamente todos los sistemas soportaron las pruebas de estrés. Solamente el sistema A9 no superó la prueba de centrifugación, en consecuencia, las pruebas de ciclado no se determinaron. La estabilidad física y termodinámica de la formulación es muy importante, ya que, si la formulación presenta alguna inestabilidad física puede haber una separación de las fases de los excipientes o presentarse la precipitación del fármaco, lo cual afectaría a su desempeño, impactando en su biodisponibilidad y por lo tanto en la eficacia terapéutica [38]. En la prueba de estabilidad física, los SEDDS deben ser capaces de resistir condiciones de estrés y mantener sus propiedades a diferentes condiciones [4].
Tabla 4 Estabilidad física de los sistemas cargados con indometacina sometidos a pruebas de estrés.
| Tratamiento | Centrifugación | Enfriamiento-calentamiento | Congelación-descongelación | ||||||||||||
| Sistema | A | B | C | D | E | A | B | C | D | E | A | B | C | D | E |
| 1 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 3 | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND |
| 4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 5 | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND |
| 6 | ND | ✓ | ✓ | ND | ✓ | ND | ✓ | ✓ | ND | ✓ | ND | ✓ | ✓ | ND | ✓ |
| 7 | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND | ND | ✓ | ✓ | ND | ND |
| 8 | ND | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ |
| 9 | x | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ |
| 10 | ND | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ | ND | ✓ | ✓ | ✓ | ✓ |
✓: estable, ND: no determinado, x: inestable.
Distribución de tamaño de partícula, índice de polidispersidad y potencial zeta
La distribución del tamaño de la partícula de los SEDDS es un factor importante porque un tamaño de partícula muy pequeño reduce la tensión interfacial del sistema, lo que promueve la absorción y biodisponibilidad del fármaco en la formulación. La tabla 5 muestra los valores de los tamaños obtenidos para los sistemas cargados con indometacina. Los valores logrados para los sistemas son menores a los 300 nm, lo cual conlleva a sugerir que tendrán una liberación más rápida del fármaco en un medio acuoso [1, 4, 6, 20]. Los valores del índice de polidispersión por espectroscopía de correlación de fotones menores a 0,3 se consideran como óptimos. Los valores resultantes para los diferentes sistemas también se muestran en la tabla 5. Los sistemas que fueron estables durante las pruebas de estrés presentan valores de índice de polidispersión menores a 0,3, lo que representa la uniformidad de la distribución de las poblaciones de glóbulos y sus tamaños [37]. Por otro lado, el potencial zeta de los sistemas que se muestra en la tabla 5 indica que las fuerzas de repulsión son lo suficientes fuertes para mantener la dispersión estable físicamente, al menos lo suficiente para mantener el sistema estable en tracto gastrointestinal. La tabla 6 muestra los sistemas que fueron estables físicamente, así como el resto de los resultados de caracterización. De los 50 sistemas propuestos inicialmente, sólo 15 de ellos fueron candidatos para comparar su desempeño con respecto al producto de referencia.
Tabla 5 Tamano de particula, indice de polidispersion (IP) y potencial zeta (ζ) de los sistemas cargados con indometacina.
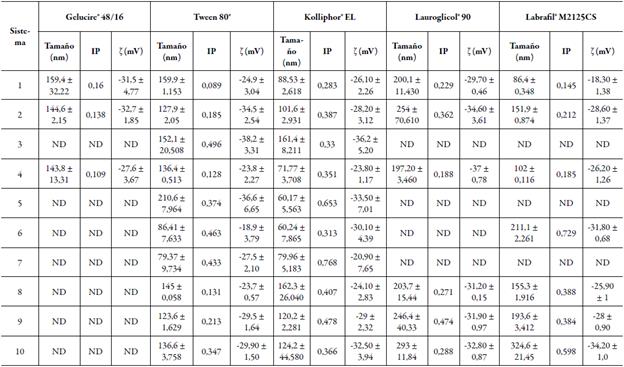
Cada valor representa la media ± D. E. (n=3).
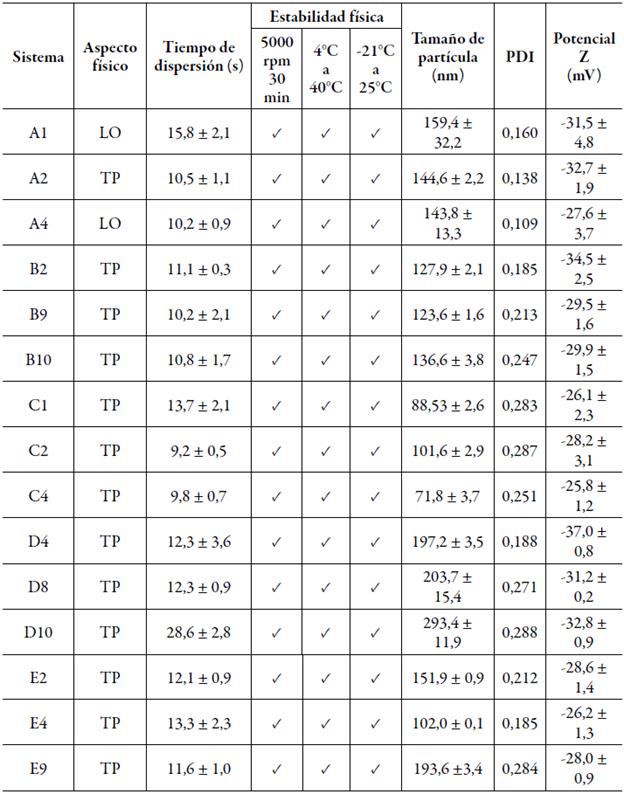
Perfiles de disolución
Los perfiles de disolución de Indocid®, que corresponde al producto de referencia para indometacina establecido en México por la Comisión Federal para la Protección contra Riesgos Sanitarios (Cofepris), y las 15 formulaciones que cumplieron con las pruebas de caracterización mencionadas en la tabla 6 se realizaron con el fin de comparar el proceso de disolución y determinar la o las formulaciones que se asemejan más al medicamento de referencia a partir de los valores obtenidos de factor de similitud f2 [23]. En la figura 6 se muestran únicamente las formulaciones que tuvieron un valor mayor que 50, las cuales corresponden a los sistemas A2, Cl y D4.
La formulación A2 corresponde al sistema que contiene 50% de Labrasol, 45% de Transcutol HP y 5% de Gelucire 48/16, la formulación Cl corresponde al sistema que contiene 90% de Labrasol, 5% de Transcutol HP y 5% de Kolliphor El y la formulación D4 corresponde al sistema que contiene 70% de Labrasol, 25% de Transcutol HP y 5% de Lauroglicol 90.
Estas formulaciones tuvieron un valor de f2 mayor a 50, presentaron un ligero aumento en la velocidad de disolución de indometacina de 0 a 10 minutos y, además, tuvieron la capacidad de presentar el fármaco en un estado solubilizado para favorecer una permeación en tiempos menores, este incremento en la velocidad de disolución es el resultado de la eliminación de las fuerzas intermoleculares presentes en el estado sólido inicial del fármaco en una forma farmacéutica sólida, ya que los SEDDS pueden presentar al fármaco previamente solubilizado en la formulación lipídica, adicionalmente, se incrementa la fuerza de interacción entre el soluto y el medio acuoso mediante el empleo de excipientes con una polaridad menor a la del agua y más a la del fármaco [8, 18, 33].
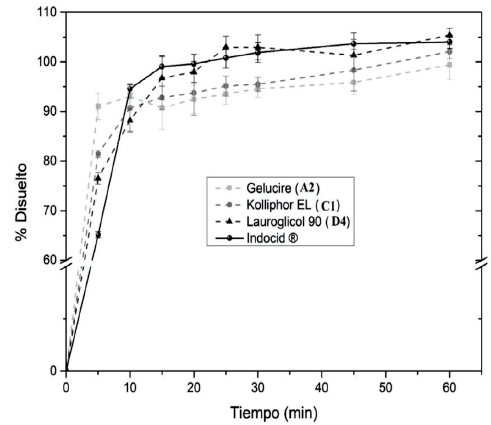
Figura 6 Perfiles de disolución de las formulaciones que presentan similitud con el medicamento de referencia Indocid®.
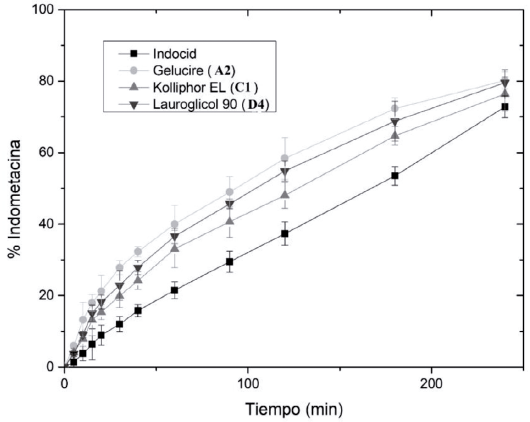
Los perfiles de difusión en membranas sintéticas representados en la figura 7 mostraron que las formulaciones SEDDS lograron liberar porcentajes de indometacina de 76 a 80% al término de las 4 h, mientras que el medicamento de referencia alcanzó un 72%, si bien estos porcentajes son muy cercanos entre sí, el porcentaje liberado de 0 a 180 minutos mostró que las formulaciones SEDDS tienen la capacidad de aumentar la velocidad de liberación del fármaco, fenómeno que se relaciona con el aumento en la velocidad de disolución de las formulaciones. Estos resultados fortalecen la hipótesis de que posterior al inicio de la formación in situ de la emulsión en los fluidos gastrointestinales [14, 15] se verá aumentada la velocidad de disolución, la permeación a través del epitelio intestinal y su posterior absorción, proporcionando así un entorno satisfactorio para la mejora de la biodisponibilidad oral de la indometacina [14, 33, 37].
CONCLUSIONES
A través de la determinación del incremento de la solubilidad con respecto al agua de los fármacos poco solubles como la indometacina es factible obtener SEDDS potencialmente útiles para la administración oral, utilizando excipientes lipídicos, así como una adecuada mezcla de ellos, conseguida mediante el diseño experimental.
El uso de los excipientes lipídicos permitió aumentar la solubilidad acuosa de la indometacina en un orden de 105 veces, así como el uso de los SEDDS permitió generar diferentes formulaciones que permiten solubilizar la dosis de indometacina.
De acuerdo con su desempeño in vitro, las formulaciones SEDDS presentaron tiempos de autoemulsificación menores a 30 segundos, además, una alta estabilidad física al ser sometidas a diferentes pruebas de estrés, el análisis de tamaño de partícula mostró diámetros hidrodinámicos inferiores a los 300 nm y con un índice de polidispersión menor a 0,3, indicativo de una alta homogeneidad en la población de partículas, así como por sus valores de potencial zeta menores de -25 mV, hecho que les confiere a los sistemas estabilidad de tipo electrostático.
Las formulaciones SEDDS A2, C1 y D4 cumplieron con la especificación farmaco-péica de tener un porcentaje disuelto de fármaco mayor al 80% en menos de 15 minutos, además de obtener un factor de similitud mayor que 50 (indicativo de un perfil de disolución del activo similar al medicamento de referencia), los sistemas A2 y Cl corresponden a los sistemas tipo IV y la formulación D4 corresponde a un sistema tipo IIIB, según la clasificación de los sistemas lipídicos de Pouton. Las 3 formulaciones presentaron un porcentaje de fármaco liberado similar al medicamento de referencia al término de 4 h, pero con un incremento notorio en la velocidad de liberación.
El incremento de la velocidad de disolución y de liberación de la indometacina in vitro refleja las ventajas de los sistemas SEDDS frente al medicamento de referencia, con una posible mejora en el tiempo de inicio del efecto terapéutico, factor de gran impacto en los medicamentos con actividad analgésica.

