











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
Cuando se habla de "química computacional" inmediatamente se piensa en el uso de supercomputadores y programas inteligentes que ayudan al investigador a encontrar una respuesta sobre un problema relacionado con la química experimental [1]. Pero no todo es tan sencillo desde el mismo concepto, si se revisa la definición de química computacional se encuentra que muchos autores la definen como la capacidad de "predicción" de propiedades químicas y físicas de sistemas atómicos o moleculares a través de programas especializados, pero el término predicción no es coherente en las ciencias exactas. Es aquí donde se debe revisar otras definiciones cambiando la incerteza de la predicción por la habilidad de "computar", "calcular" o "estimar" dichas propiedades [2]. Estos debates siempre han sido parte de la historia de la química al definir una nueva área, por ejemplo, la fina línea que existe entre la definición de la química orgánica e inorgánica.
En la actualidad la química computacional es muy importante para el desarrollo y el descubrimiento de nuevas propiedades químicas, como por ejemplo el recientemente descubierto "enlace vibracional" [3], el diseño y descubrimiento de medicamentos [4], el estudio de mecanismos de reacciones químicas [5], la estimación de propiedades termodinámicas [6] y la energía de correlación electrónica de extensos y complejos sistemas moleculares de tipo orgánico [7], la simulación de sistemas biológicos [8], e inclusive aporta información para el entendimiento de la evolución química del universo [9]. Todas estas investigaciones se desarrollan realizando cálculos físico-matemáticos de diversos tipos como métodos de elemento finito (macro escala), cálculos de campos de fuerza (meso escala), y cálculos de primeros principios (ab initio) que buscan describir la estructura electrónica de átomos y moléculas [10], ver figura 1.
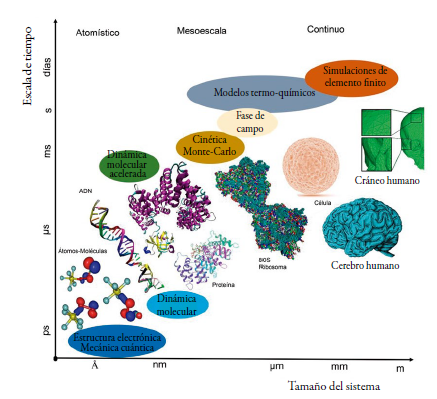
Figura 1 Clasificación aproximada de los métodos de simulación en diferentes escalas de tiempo y tamaño.
Los cálculos ab initio intentan resolver matemáticamente las ecuaciones de la mecánica cuántica HΨ = EΨ para un sistema atómico y molecular sin usar ningún método empírico o entradas experimentales [11], la muy conocida teoría funcional de la densidad (DFT) pertenece a esta familia [12] (aunque algunos investigadores están en desacuerdo debido a que algunos funcionales incluyen parámetros experimentales o empíricos en sus formalismos) junto a las teorías Hartree-Fock (HF) y Post Hartree-Fock [13]. En términos de exactitud y costo, las teorías comúnmente empleadas son HF, la teoría Moller-Plesset (MPn, n=1,2,3...) y la teoría Coupled-Cluster (CC) [14] en combinación con complejos conjuntos de bases que describen los orbitales atómicos y moleculares. Este tipo de cálculo y los fundamentos de la termodinámica estadística han desarrollado un gran campo de aplicación denominada "termoquímica computacional". Esta permite estimar directamente las energías de entalpías de reacción (ΔHr), entalpías de formación (ΔHf), entalpías de sublimación (ΔHsub), entalpías asociadas a procesos de afinidad electrónica (AHAE), energías libres de Gibbs (ΔG° y ΔG‡), energías atomización (ΔD0), entre otras [15].
METODOLOGÍA EN TERMOQUÍMICA COMPUTACIONAL
El uso de supercomputadoras para realizar estudios de termoquímica computacional empleando complejos cálculos ab initio del tipo interacción de configuración completa (del inglés Full CI) son una limitación importante para el estudio de sistemas moleculares de pequeño y mediano tamaño debido a que estos proporcionan soluciones numéricas exactas de la ecuación electrónica no relativista de Schrödinger independiente del tiempo [16]. Para superar esta limitación y acercarse a la deseada precisión química de 1,0 kcal/mol (4,184 kJ/mol) se han desarrollado métodos conformados por una serie de cálculos con diferentes funciones de base y niveles de teoría denominados métodos compuestos, estos permiten obtener energías muy próximas al que se obtendría mediante un cálculo del tipo Full CI pero con un menor costo computacional [17].
Los métodos compuestos más utilizados son: La serie Petersson de base completa (CBS) [18], las teorías Gaussian-n [19], los métodos Weizmann-n [20], el modelo HEAT (del inglés high-accuracy extrapolated ab initio thermochemistry) [21], el procedimiento FPD (en honor a sus desarrolladores Feller-Peterson-Dixon) [22], entre otros, que además se pueden combinar con otras estrategias como los esquemas de reacciones (isodésmicas e isogíricas) [15], correcciones de aditividad de enlace BAC [23, 24], aditividad de grupos [25], etc., para ayudar a mejorar las energías estimadas.
Las series Petersson CBS como CBS-CI, CBS-2, CBS-APNO, CBS-4, CBS-QB3, etc., involucran esencialmente siete u ocho pasos (ver figura 2), un primer paso de optimización de la geometría y posteriormente sucesivos cálculos de punto simple con largos conjuntos de base, que de forma aditiva corrigen la energía de la función de onda de partida.
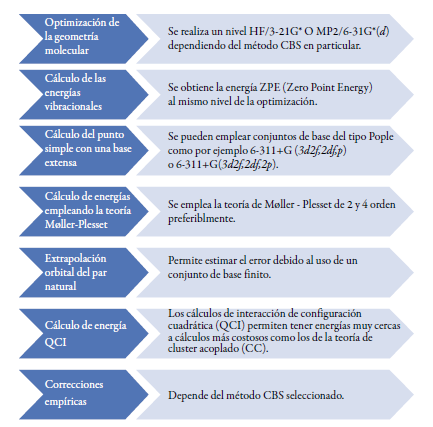
Se realiza un nivel HF/3-21G* O MP2/6-31G*(á) dependiendo del método CBS en particular.
Las teorías Gaussian-w han sido desarrolladas desde los años 90 y sus últimos desarrollos corresponden a las teorías G4 [26] y G4MP2 [27] las cuales están incluidas en el programa Gaussian en sus versiones 09 y 16. La teoría G4 presenta una desviación media absoluta de 0,83 kcal/mol para 454 energías experimentales compuestas por entalpías de formación, potenciales de ionización, afinidades electrónicas, protónicas y energías de enlaces de hidrógeno evaluadas.
La teoría G4 consiste en varios pasos, el primero de ellos (1) consiste encontrar la geometría de equilibrio a un nivel B3LYP/6-31 G(2df,p), seguido de un cálculo de energía con su respectiva corrección del punto cero E(ZPE) escalando las frecuencias obtenidas a este nivel de teoría con un factor de 0,9854, para obtener E0; (2) un procedimiento de extrapolación obtenido a partir de un cálculo Hartree-Fock limite E(HF/limit) para su inclusión en el cálculo de la energía total con un incremento en el conjunto de polarización de d a 3d sobre los átomos de la primera fila y Ad sobre los átomos de la segunda fila con reoptimización de los exponentes del conjunto para 4d como se muestra en la ecuación 1; (3) un cálculo de energía del punto simple empleando teorías perturbativas (MPn) y acopladas CCSD(T); (4) dos nuevos parámetros para la corrección de alto nivel (HLC) que tienen en cuenta las deiciencias de los radicales y de especies con un solo par de electrones en la capa de valencia.
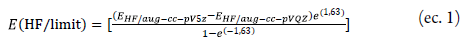
El cálculo de la energía E(combined) es la sumatoria de todas las correcciones de los valores de las energías del punto simple mencionadas anteriormente y se expresa mediante la ecuación 2:
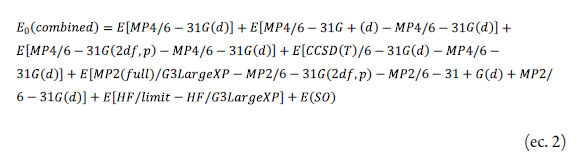
La energía total E0(G4) es la suma de E(combined), E(ZPE) y la corrección de alto nivel (HLC). La corrección de alto nivel (HLC) está dada por las expresiones -Anß para moléculas de capa cerradas, -A'nß -B(na -nß) para sistemas de capa abierta y -Cnß -D(na -nß) para átomos (incluyendo iones). Además, se puede adicionar el parámetro E, este corrige las energías de pares de electrones en especies moleculares y atómicas donde los electrones de valencia consisten en solo un par de electrones s (no incluye sistemas que tienen uno o más electrones 1s). Los términos A, A', B, C, D y E en hartrees para el método G4 son A=6,947x10-3, A'=7,128x10-3, B=2,441x10-3, C=7,116x10-3, D=1,414x10-3 y E= 2,745x10-3.
El método G4(MP2) es una variación de la teoría G4 con menor costo computacional al reemplazar los cálculos MP3 y MP4 de la teoría de perturbaciones Moller-Plesset por cálculos MP2 con bases más extensas. G4(MP2) optimiza las geometrías al mismo nivel que G4. Este método inicia con un cálculo de energía simple que se realiza a un nivel de teoría CCSD(T) con un conjunto de base 6-31G(d), luego esta energía es mejorada con una serie de correcciones de energía y solo los electrones de valencia son correlacionados. La ecuación (3) denota la energía total E 0 , FC denotafrozen core.
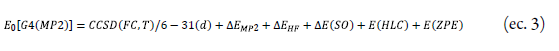
La corrección de las energías MP2 se presenta en la ecuación 4:
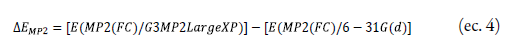
En la ecuación 4 se observa un conjunto de base G3MP2LargeXP que es una variación de la base G3LargeXP en G4. El termino AEHF = E[HF/limit - HF/G3MP2LargeXP] y las correcciones HLC fueron obtenidas de forma similar que en la teoría G4. Los términos A, A', B, C, D y E en hartrees para el método G4(MP2) son A=9,472x10-3, A'=9,769x10-3, B=3,102x10-3, C=9,741x10-3, D=2,115x10-3 y E= 2,379x10-3 [28]. La desviación media absoluta de este método es de 1,03 kcal/mol.
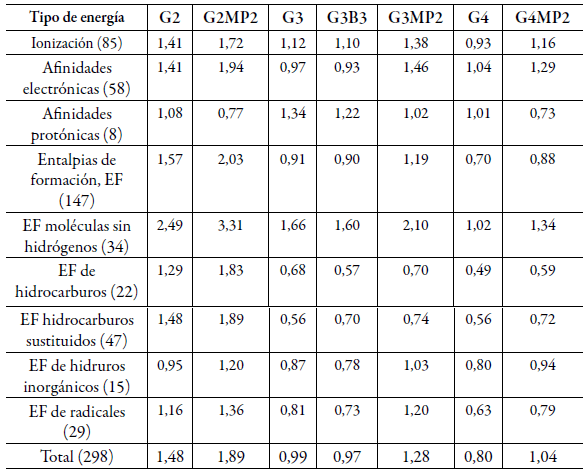
En la tabla 1 se puede observar la mejora de los métodos compuestos G4 y G4MP2 respecto a sus versiones anteriores G3(MP2), G3//B3LYP, G3, G2(MP2) y G2 en la estimación de energías de ionización, ainidades electrónicas, ainidades protónicas y entalpias de formación al compararlas con un conjunto de prueba de valores experimentales de 298 moléculas denominado G2/97 [19].
Tabla 1 Desviación media absoluta (en kcal/mol) de la comparación de energías de varios métodos Gn y valores experimentales del conjunto de prueba G2/97*. En paréntesis se muestra la cantidad de moléculas evaluadas. *Tomado de referencia [19].
Las teorías Weizmann-n emplean cálculos ab initio con largos conjuntos de base como, por ejemplo, el llamado W1 incluye un cálculo de punto simple al nivel teórico CCSD(T)/aug-cc-pVQZ+2D1f y es capaz de estimar errores de 1,01 kcal/mol en comparación con los 0,64 obtenidos a partir de W2. Ambas propuestas son útiles para el estudio de sistemas moleculares de pequeño tamaño o moléculas de no más de 10 átomos pesados [18], se destacan por no incluir parámetros empíricos e incluir efectos relativistas. Esta misma idea estratégica pero con conjuntos de bases extensos tipo Dunning [29] al nivel teórico de clúster acoplados (CC) es empleada en el modelo HEAT, el procedimiento FPD y otros métodos compuestos.
APLICACIONES EN TERMOQUÍMICA COMPUTACIONAL
Termoquímica de nuevos fármacos
Muchos compuestos heterocíclicos han demostrado tener actividad biológica del tipo antibacteriana, anticancerígena, antiinflamatoria y anticonvulsiva, inclusive se ha utilizado farmacológicamente para el tratamiento en enfermedades neurológicas y crónicas como el Alzheimer y la artritis reumatoide. [30] Sin embargo, comprender la reactividad, el comportamiento de los enlaces y las condiciones estructurales de estos compuestos es de gran interés para la industria farmacéutica, presenta un reto para la química debido a que los datos termoquímicos experimentales con alta precisión son escasos y asignados a un reducido número de compuestos. Moyassar y colaboradores [30] estudiaron 34 especies químicas correspondientes a hidroxiquinolinas, sus ceto-tautómeros derivados y los análogos de aza-azuleno. En su metodología fueron estimadas las entalpias de formación en fase gaseosa, las energías de ionización (IE) y las ainidades electrónicas (EA) empleando la serie Petersson de base completa CBS-QB3. Sus resultados de las entalpias de formación en comparación con las entalpias experimentales de los compuestos fenol, 2-hidroxipiridina y 4-hidroxipiridina (ver figura 3) presentaron una alta precisión con un error estándar medio de 5,2 kJ/mol. Para el caso del azuleno la entalpía de formación experimental reportada por varios autores fue de 280,0 kJ/mol, 308,0 kJ/mol y 288,0 (±5,3) kJ/mol pero al emplear los métodos compuestos G3 y G4 los valores estimados para el azuleno son de 295,0 y 299,0 kJ/ mol, respectivamente. Lo anterior muestra la precisión química de la termoquímica computacional [30].
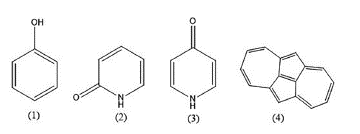
Figura 3 Compuestos químicos estudiados con el método compuesto CBS-QB3 (1) Fenol, (2) 2-hidroxipiridina, (3) 4-hidroxipiridina, (4) Azuleno.
Otro grupo de fármacos muy conocidos derivados del ácido N-fenilantranílico son los fenamatos, estos hacen parte del grupo de antiinflamatorios no esteroideos que han sido utilizados para el tratamiento de la artritis reumatoidea y la osteoartritis [31], poseen propiedades antiinflamatorias, antipiréticas y analgésicas. A pesar de tener variedad de beneficios, son compuestos poco estudiados termoquímicamente. Surov y colaboradores [32] estudiaron las propiedades termoquímicas de la difenilamina y el ácido N-fenilantranílico que corresponden a compuestos precursores de los fenamatos. El estudio permitió la determinación de entalpías molares de formación que se obtuvieron por calorimetría de combustión, donde se reporta por primera vez un valor de 118,5 (±2,9) kJ/mol para el ácido fenilantranílico. Las entalpías de sublimación se obtuvieron a partir de los resultados de presión de vapor obtenidos por el método de transpiración reportando valores de 95,2 (±0,6) kJ/mol para la dimetilamina y 126,0 (±1,3) kJ/mol para el ácido N-fenilantranílico a 298,15 K.
Se estimaron valores de entalpias de formación en fase gaseosa correspondientes a 206,2 kJ/mol mediante el método compuesto G3(MP2) frente a un valor experimental de 213,7 kJ/mol para la dimetilamina y -187,4 kJ/mol frente a un valor experimental de -180,0 kJ/mol para el ácido N-fenilantranílico (ver figura 4) [32]. En este trabajo se logró comprobar la consistencia interna de los datos sobre las propiedades termoquímicas y permitió la validación y coherencia de los datos experimentales con los valores estimados por métodos cuánticos modernos.
Los estudios termoquímicos han permitido realizar análisis y mejoras de los mecanismos de desintoxicación de algunos fármacos aumentando su efectividad. Sahu y colaboradores [33] realizaron un estudio computacional basado en mejorar los mecanismo de desintoxicación con el cual actúa el fármaco antilewista británica (BAL 2,3 dimer-captopropanol) como antídoto contra el envenenamiento por arsénico utilizado en la segunda guerra mundial y actualmente empleado para el tratamiento por intoxicación de metales pesados. El mecanismo de desintoxicación de este fármaco consiste en la ruptura del enlace As-Cl para dar la formación de enlaces As-S produciendo un producto estable y no tóxico que inalmente es excretado. Para esto, se realizaron cálculos al nivel de teoría M06-2X/TZVP y M06-2X/cc-pVTZ-PP para establecer la energética del mecanismo de desintoxicación en presencia de catalizadores de NH3 y H2O tanto en fases gaseosas como en tres tipos de solventes: el ciclohexano, dimetilsulfóxido y agua. [33] En esta investigación se empleó la teoría del estado de transición (TST) para estimar las constantes de velocidad de las reacciones catalizadas para el H2O y para el NH3, los valores reportados de 2,42x10-11 s-1 y 2,88x10-11 s-1 respectivamente, muestran que estas reacciones son razonablemente más rápidas que la desintoxicación no catalizada de 5,44x10-13 s-1. Esta investigación computacional se convierte en una base clave para la posterior investigación experimental para entender o mejorar el proceso de quelación del arsénico u otros metales en presencia de fármacos que contienen aminoácidos básicos o grupos hidroxilo como es el caso del dimercaprol (Antilewista Británica, BAL 2,3 dimercaptopropanol).
Química de la atmósfera
En la atmósfera una gran variedad de especies químicas son contaminantes de origen antropogénico los cuales afectan sus procesos dinámicos y logran desequilibrar su composición natural, esta situación se ha descontrolado en los últimos años y se ha evidenciado en fenómenos ambientales como las lluvias ácidas [34], el efecto invernadero y el cambio climático [35]. Estas reacciones atmosféricas son difíciles de investigar a nivel de laboratorio debido a que muchas de estas especies son muy inestables y tienen tiempo de vida muy cortos [36]. Los principales contaminantes atmosféricos son los óxidos de carbono, nitrógeno y azufre (COx, NOx y SOx), además de diversos compuestos orgánicos volátiles (COV), la gran familia de los clorofluorocarburos (CFC), y el hexafluoruro de azufre S F6 según lo registrado en la lista del Intergovernmental Panel on Climate Change (IPCC) sobre gases de efecto invernadero [37].
En los últimos años ha investigado las características estructurales, espectroscópicas, energéticas y cinéticas de varias especies de relevancia real o potencial en la química de la atmósfera empleando diversos funcionales de la teoría del funcional de la densidad (DFT) y métodos compuestos ab initio. Derivados del hexafluoruro de azufre (SF6) son considerados los nuevos gases de efecto invernadero y la gran alarma sobre estos compuestos se dio por la detección en la atmósfera del nuevo gas de efecto invernadero penta fluoro-sulfuro de trifluoro metilo SF5CF3 en el año 2000 [38] el cual tiene un potencial de calentamiento global (GWP) de 17.700 estimado a 100 años en relación con el CO2 [39]. Con este antecedente de importancia ambiental se investigó la serie de compuestos SFxCl (x=1-5) y SF5On (n=1-3) a través de cálculos de la química cuántica ya que la información experimental es muy poca e inexacta, en la tabla 2 se muestran valores de entalpías de formación de la serie SFxCl (x=1-5).
Tabla 2 Entalpías de formación (kcal/mol) calculadas a 298 K para la serie SFxCl (x=0-5) a partir de energías de atomización totales.
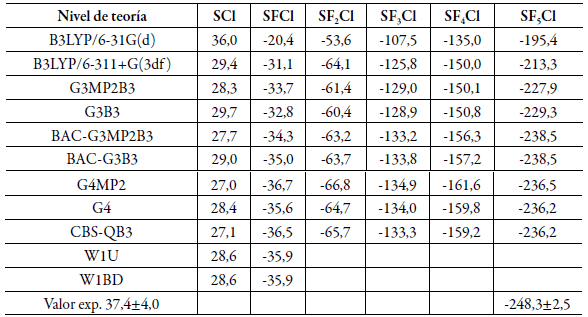
En la literatura el valor experimental aceptado para el radical SCl a 298 K es 37,4±4,0 kcal/ mol y este difiere con el valor de 28,6 kcal/mol estimado por los métodos W1U y W1BD. Sin embargo, existe una coincidencia totalmente fortuita con el valor estimado al nivel teórico B3LYP/6-31G(d) que se hace menos confiable debido a que al aumentar la calidad de la base, la entalpía de formación que se deriva es notablemente menor. Un comportamiento similar se observa con el valor para la entalpía de formación del SF5Cl donde hay una diferencia de 12 kcal/mol entre el valor experimental aceptado y el estimado con los métodos G4 y G4MP2. En general, las entalpías de formación de las moléculas de la serie SFxCl tienden a ser menores a medida que aumenta el número de átomos de flúor y es recomendable revisar los experimentos con los cuales se estimaron los valores de entalpías para algunos compuestos de la serie [40].
Los radicales SF5O, SF5O2 y SF5O3 han sido objeto de estudio por varios investigadores como subproductos de la disociación del peróxido y trióxido de bis-penta-fluoruro de azufre SF5OOSF5 y SF5OOOSF5 [41-43] Kronberg y colaboradores [44] sintetizaron estos radicales a nivel de laboratorio y emplearon el nivel teórico B3LYP/6-311++G(3df,3pd) para el estudio de las estructuras de estos radicales. Sin embargo, solo este nivel teórico empleado es ineiciente para reportar información estructural y energética adecuada para los radicales debido a la alta contaminación de spin, por lo que estos radicales fueron reevaluados por Xu y colaboradores [45] con diversos funcionales DFT y empleando el conjunto de base DPZ (double-Z plus polarization). En este estudio se reportó una diferencia del grupo puntual para el radical trióxido SF5OOO de C1 frente a la Cs reportada por Kronberg. Recientemente, este radical SF5OOO fue estudiado empleando diversos funcionales DFT y su energética a través de métodos compuestos [46], Buendía y col. reportaron una nueva conformación geométrica Cs' para este radical y sus valores de entalpías de formación son de -218,3 ± 1,3, -217,2 ± 1,6 y -218,0 ± 1,5 kcal/mol para las conformaciones Cs, Cs' y C1 a 298 K, respectivamente, estimados a partir de los métodos G3MP2B3, G3B3, G4MP2 y G4.
Derivados del petróleo
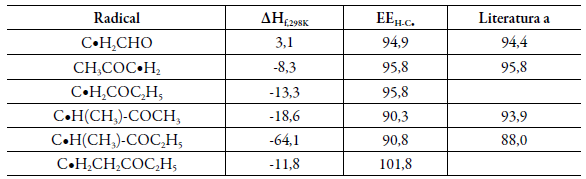
La formación, conversión y transporte de especies químicas derivadas del petróleo dentro de los motores de combustión son de especial interés no solo por su aplicación industrial sino por su comportamiento químico en general. Algunos combustibles derivados han sido empleados en motores con el fin de hacerlos más eficientes y mejorar procesos, sin embargo, su comportamiento químico es desconocido en atmósferas de oxidación y consecuentemente no ha sido posible entender los rangos de temperatura y presión en los cuales estos compuestos son útiles. Para conocer dicho comportamiento es necesario definir las propiedades termoquímicas del sistema, con alta precisión, y en vista de la dificultad de obtenerlas bajo procedimientos experimentales, la química computacional y especíicamente los métodos compuestos aportan gran información. Se ha empleado el método CBS-QB3 para obtener la entalpía de formación del combustible traza radical 2-Oxopropil o Acetonil (CH3COC●H2) [47] estimándose un valor de -8,3 kcal/mol frente al valor experimental reportado por Bouchoux [48]. La importancia de este valor estimado es que a partir de esquemas de reacciones iso-désmicas se pueden estimar los valores de entalpía de formación para la familia completa de radicales de la acetona, 2-oxoetilo o formilmetilo (C●H2CHO), 2-oxobutilo (C●H2COC2H5), 1-metil-2-oxopropilo o metilacetonilo (C●H(CH3)COCH3), 1-metil-2-oxobutilo (C●H(CH3)COC2H5), y 3-oxopentilo (C●H2CH2COC2H5). La tabla 3 muestra las energías de enlace entre el carbono radical y su respectivo átomo de hidrogeno donde se puede apreciar una diferencia de menos de 4,8 kcal/mol frente a los datos experimentales conocidos.
Tabla 3 Entalpías de formación a 298 K (AHf298K) y Energías de enlace H-C» (EEH-C») para los radicales derivados de la acetona en kcal/mol.
a Datos reportados por El-Nahas et al. [48].
Otras investigaciones no solo buscan entender el comportamiento químico de combustibles derivados del petróleo sino también utilizan estos métodos ab initio para buscar nuevas especies que sean limpias con el medio ambiente. Con esta finalidad dos diferentes familias de hidrocarbonos oxigenados (alcoholes, aldehídos, ácidos carboxílicos, etc.) y ésteres de metilo candidatas para producir biodiesel han sido estudiadas [49] mediante la determinación de sus energías de disociación de enlace, entalpías de reacción y constantes cinéticas de reacción dependientes de presión y temperatura a través de teorías de multireferencia de estructura electrónica de interacción simple y doble (MRSDCI) [50]. Los alcoholes etanol y metanol estudiados bajo el método mencionado presentan una entalpía de disociación para todos los casos en un rango de 83,2-102,7 kcal/mol, con un error de 1,7 kcal/mol frente a los datos experimentales reportados. En el caso de aldehídos, ácidos y ésteres como el formaldehido, acetona y ácido acético se estimaron valores de entalpía de disociación un rango entre 81,1-97,0 kcal/mol con un error de 1,0 kcal/mol frente a los datos experimentales [49].
Por su parte, valores precisos de energías de disociación de enlace (BDE) para cadenas hidrocarbonadas de 16 a 18 átomos fueron determinados para la segunda familia de ésteres metílicos de alto interés como biodiesel. Particularmente los ésteres (C1-C4) for-miato de metilo, acetato de metilo, propanoato de metilo y metil-butanoato de metilo, presentan entalpías de disociación de enlace en un rango de 85,1-103,1 kcal/mol. Ésteres de cadena más larga como el estearato de metilo, oleato de metilo, linoleato de metilo y linolenato de metilo presentan entalpías de disociación de enlace en un rango similar a los ésteres de cadena corta, 81,6-109,1 kcal/mol. Con esta información se logró concluir que los enlaces C=C son más débiles en los ésteres alquílicos y probablemente son los sitios en los que los combustibles pirolizan a altas temperaturas de combustión [49].
Diseños de nuevos explosivos
El diseño de explosivos ha sido un atractivo para los químicos computacionales que buscan encontrar respuestas a la sensibilidad y naturaleza de los mismos. De esta manera, estudios de descomposición térmica de diferentes familias de compuestos se han llevado a cabo utilizando métodos compuestos G2, G3, G3B3 y CBS-QB3. Por ejemplo, algunos derivados de compuestos nitro aromáticos que poseen un fuerte enlace C-NO2 son de gran interés para este campo de investigación y han sido caracterizados por diversos métodos ab initio. Los valores de entalpía de formación permiten comparar los efectos que distintos sustituyentes presentan sobre la fuerza del explosivo. Un total de 28 especies, Nitroben-ceno, o-Nitroanilina, Ácido o-Nitrobenzoico, o-Fluouronitrobenceno, m-Nitroanilina, Ácido m-Nitrobenzoico, m-Fluouronitrobenceno, p-Nitroanilina, Ácido p-Nitroben-zoico, p-Fluouronitrobenceno, o-Nitrotolueno, o-Dinitrobenceno, o-Cloronitroben-ceno, m-Nitrotolueno, m-Dinitrobenceno, m-Cloronitrobenceno, p-Nitrotolueno, p-Dinitrobenceno, p-Cloronitrobenceno, o-Nitrobenzaldehido, o-Nitrobenzonitrilo, o-Nitrofenol, m-Nitrobenzaldehido, m-Nitrobenzonitrilo, m-Nitrofenol, p-Nitroben-zaldehido, p-Nitrobenzonitrilo y p-Nitrofenol, fueron estudiadas presentando valores de desviación promedio absoluta de 1,2, 1,1, 1,6 y 2,9 kcal/mol, para los métodos G2, G3, G3B3 y CBS-QB3, respectivamente en la estimación de sus entalpías de formación frente a datos experimentales [51].
Debido a que un compuesto explosivo contiene una gran energía en sus enlaces, algunas especies químicas, no solo tienen utilidad en esta área sino también son blancos para el desarrollo de combustible de cohetes. Es por ello que la molécula Carbonil Dia-zida (CN6O) [52] fue reevaluada con el fin de determinar si este compuesto presenta un peril característico como explosivo y si la energía contenida puede ser conducida al desarrollo de combustibles de alta energía. El método G3B3 fue utilizado en el estudio y particularmente el isómero OCN4-N2 presento una barrera de disociación es 30 kcal/mol y un impulso especifico (252,3-270,7s) cercano a los explosivos RDX (Ciclotrime-tilen trinitroamina) y HMX (Ciclotetrametilen trinitroamina).
Una familia de compuestos hidro-nitrogenados N4H4 formada por once isómeros divididos en tres grupos cliclotetrazenos, ciclotriazenos y tetrazenos de cadena recta son de gran relevancia por la energía contenida en sus enlaces ya que poseen el grupo químico hidracina que es bien conocida en sus aplicaciones como combustible y propelente. La termoquímica de esta familia ha sido estudiada por el método G3B3 [53] y los valores estimados muestran que los calores de formación a 298 K están en un rango de 301,3602,8 kcal/mol, resultado que es positivo para que estas moléculas sean candidatas en aplicaciones como explosivos. El compuesto NH2-N=N-NH2 fue el más estable y de forma particular las estructuras cíclicas de cuatro miembros son menos estables que los cíclicos de tres miembros pese a la tensión de estas estructuras, esta característica debe ser estudiada a mayor profundidad.
El desarrollo de materiales de alta energía se ha enfocado en estudiar los compuestos hidrocarbonados tetraedranos y cubanos nitro-sustituidos para revisar su capacidad explosiva. Un total de 22 especies (ver figura 5) fueron evaluadas por Reyne y Forest con los métodos de cálculo G4MP2 y G4 [54].
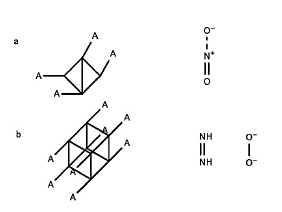
Figura 5 (a) Compuestos tetraedranos y sus derivados (nitro-NO2). (b) Compuestos cubanos y sus derivados (azo (N=N), nitro y peroxo (O-O)) [41].
Sus entalpías de combustión ΔH°comb(g) reportadas muestran que los tetraedranos tienen de exotermicidad en un rango de -2,3 y -9,4 kcal por gramo de compuesto, este resultado es desfavorable para emplearlo como compuestos de alta energía. Por otra parte los derivados cubanos presentaron entalpías de formación entre 130,2 y 135,2 kcal/mol valores que no diieren del cubano sin sustitución con ΔH°f,298K de 144,2 kcal/mol, solo los valores obtenidos para mono- y dinitrocubanos marcaron la diferencia y sus entalpías se comparan muy favorablemente con explosivos primarios representativos [54].
Muchos de los casos expuestos anteriormente carecen de valores experimentales por lo que es importante realizar este tipo de análisis químico de forma que se generen valores termoquímicos de partida para futuras experimentaciones que ayudan a reducir costos e impactos ambientales.
CONCLUSIONES
La termoquímica computacional es una herramienta actual para resolver problemas de la química donde la experimentación es difícil y con un alto costo económico. En la última década se han desarrollado métodos teóricos que permiten estimar energías dentro de la conocida precisión química (~ 1,0 kcal/mol) con el apoyo de sistemas de cómputo que cada día son más eficientes lo que permite aumentar la escala de los sistemas moleculares de interés. De los métodos compuestos expuestos se destacan las teorías Gaussian Gn por su precisión, versatilidad y disponibilidad, en este contexto se presentaron algunas de las aplicaciones modernas en el diseño y estudio de fármacos, la química de la atmósfera, estudios de compuestos derivados del petróleo y desarrollo de explosivos frente a las aplicaciones tradicionales en el campo de cinética química y los mecanismos de reacción.
Esta área de la ciencia va en constante crecimiento y se espera que en los siguientes años la termoquímica computacional desarrolle nuevos métodos y códigos computacionales que permitan estudiar sistemas moleculares de gran tamaño importantes en otras áreas de las ciencias como la física, la biología, ciencias de los materiales, entre otros.

