










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
Print version ISSN 0034-7434On-line version ISSN 2463-0225
Rev Colomb Obstet Ginecol vol.54 no.2 Bogotá Apr./June 2003
* Profesor Asociado. Director de la línea de Investigación en Bioquímica y Salud. Universidad de Caldas. Manizales. Colombia. Instituto de Bioquímica Clínica. Facultad de Medicina. Universidad Autónoma de Barcelona. España.
RESUMEN
La producción y regulación de la glucosa, así como el balance neto entre los requerimientos de cada sistema orgánico determinan las vías metabólicas requeridas en la producción de energía. Durante el embarazo normal, la glucosa y los combustibles metabólicos son suministrados al feto de una manera bien regulada. La diabetes durante el embarazo es una de las principales causas de alteración en el metabolismo materno afectando la glucorregulación y el desarrollo fetal. La presente revisión hace enfásis en el metabolismo de los carbohidratos, y los principales procesos para la producción de energía mediante el uso de estas biomoléculas durante el período gestacional.
Palabras clave: embarazo, metabolismo, carbohidratos.
SUMMARY
The production and regulation of glucose, as well as the net balance between systemic requirements, decide the necessary metabolic pathways for energy production. During normal pregnancy, the glucose and metabolic fuels are provided in a well-regulated manner. Diabetes in pregnancy is one of the principal causes of changes in maternal metabolism that affects fetal development and alters neonatal gluco-regulation. The present review is focused on carbohydrate metabolism, and the description of the main processes for energy production and its use during the gestational period.
Key words: pregnancy, metabolism, carbohydrates.
INTRODUCCIÓN
Las proteínas, los ácidos nucleicos, los lípidos y los carbohidratos son considerados los cuatro grupos mayores de biomoléculas. Los carbohidratos conforman la mayor parte de la materia orgánica en el planeta, sirviendo como reservas de energía; combustibles e intermediarios metabólicos; componentes de los ácidos nucleicos; elementos estructurales en paredes celulares de bacterias, plantas, y exoesqueletos de artrópodos. Además se encuentran combinados con proteínas y lípidos formando moléculas complejas que garantizan la vida en los organismos vivos, y están presentes en las superficies celulares para los procesos de reconocimiento célula-célula.(1) Químicamente son compuestos aldehídicos o cetónicos con múltiples grupos hidroxilo y la glucosa es el centro de todo su metabolismo, siendo la fuente universal de combustible para la célula humana y la fuente de carbono para la síntesis de muchos otros compuestos.(2) Otros azúcares de la dieta (principalmente fructosa y galactosa), son convertidos a glucosa o intermediarios del metabolismo de la glucosa, siendo esta precursor fundamental de compuestos no-carbohidratos, como lípidos (ácidos grasos, colesterol, hormonas esteroideas), aminoácidos, y ácidos nucleicos.(3) Solo aquellos compuestos sintetizados a partir de vitaminas, aminoácidos esenciales y ácidos grasos esenciales, no pueden ser sintetizados a partir de la glucosa en humanos.(4)
En la presente revisión, hacemos énfasis en su metabolismo, por lo cual se hace necesario recordar una serie de características de este gran grupo de compuestos, antes de avanzar describiendo procesos de degradación o síntesis. Temas como el metabolismo de proteínas y lípidos, gasto energético y balance de nitrógeno, cambios metabólico-hormonales, y bases funcionales placentarias han sido revisadas previamente.(4,5) Se utilizarán siglas internacionales para diferentes compuestos y enfermedades.
ABSORCIÓN Y DISTRIBUCIÓN DE AZÚCARES
Los carbohidratos de la dieta son digeridos en el intestino materno a través de enzimas pancréaticas e hidrolasas.(6) Los productos finales de la digestión son D-glucosa, D-galactosa y D-fructosa.(7) Estos monosacáridos son absorbidos por los enterocitos maduros del duodeno y yeyuno.(8) La absorción de glucosa ocurre en dos etapas: co-transporte con Na+ del lumen intestinal al enterocito con consumo de energía,(9) y transporte facilitado hacia la sangre mediante los transportadores de glucosa 2 (GLUT 2) sin incurrir en gasto de energía.(10) Los transportadores de glucosa existen en diferentes células como una familia de proteínas similares (isoformas), con un 50% a 76% de similitud en la cadena de aminoácidos.
La galactosa es absorbida utilizando los mismos mecanismos de la glucosa,(12) mientras que la fructosa ingresa y abandona las células abortivas del epitelio intestinal mediante difusión facilitada,(13) y el transportador encargado de llevarla al torrente sanguíneo es el GLUT 5, el cual puede transportar también glucosa pero muestra mayor actividad con fructosa.(14) Por razones aún desconocidas la fructosa se absorbe a mayor velocidad cuando es ingerida como sucrosa que cuando se ingiere sola,(15) a una de las razones fundamentales para evitar la sucrosa y preferir el consumo de frutas o zumos de frutas cuando se recomiendan ciertos tipos de dieta para reducción de peso.(16)
Las propiedades de los transportadores de glucosa varían dependiendo del tipo de tejido,(17) en el hígado, la Km para el transportador de glucosa es relativamente alto, comparado con el de los otros tejidos,(18) favoreciendo el flujo neto de glucosa al interior del hígado cuando la concentración de glucosa sanguínea se incrementa después de la ingesta de alimento, o la salida cuando los niveles en sangre disminuyen.(19) Cuando se tienen niveles de glucosa entre 18 y 54 mg/dL, se produce respuesta hipoglucémica, con signos característicos, los cuales son el resultado de la disminución en el aporte de glucosa al cerebro.(20)
ALMACENAMIENTO Y UTILIZACIÓN DE RESERVAS ENERGÉTICAS
Homeostasis de la glucosa
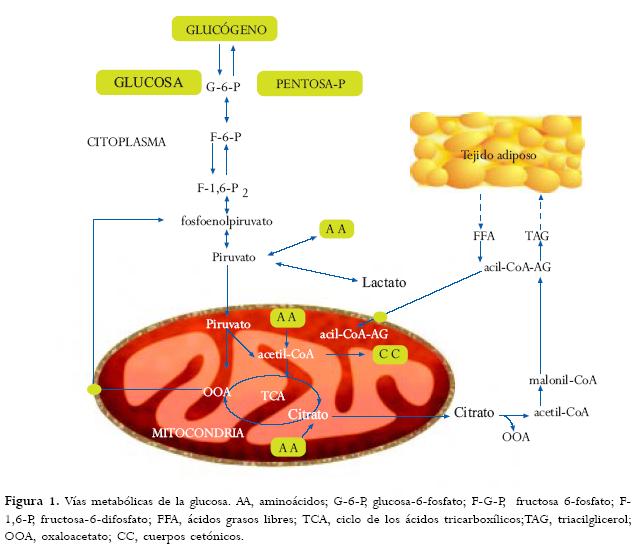
Durante la alimentación, el hígado almacena energía en forma de glucógeno y triglicéridos, estos últimos almacenados finalmente en el tejido adiposo.(21) Durante el ayuno, la glucosa y los cuerpos cetónicos son liberados.(22) El mantenimiento de un nivel de glucosa normal en sangre depende de varios factores a saber: sistemas enzimáticos glucogénicos y glucogénicos funcionales;(23) adecuado suministro de substratos gluconeogénicos aminoácidos, glicerol, lactato;(24) adecuado suministro por parte de la b-oxidación de los ácidos grasos para sintetizar glucosa y cuerpos cetónicos;(25) funcionamiento normal del sistema endocrino para integrar y regular estos procesos.(26)
La glucosa, la insulina y el glucagón son las mayores señales para controlar la transición entre la alimentación y el ayuno,(27) influenciando directa o indirectamente las enzimas que regulan el metabolismo hepático de los carbohidratos y los lípidos,(28) orientando por lo tanto, los flujos metabólicos hacia almacenamiento de energía o liberación de sustrato.(29)
Basado en el origen de la glucosa, se puede dividir la homeostasis en 3 grandes fases a saber:
1. Fase absortiva: la glucosa sanguínea se deriva principalmente de los carbohidratos exógenos durante 3 ó 4 horas después de la ingestión de alimentos.(30) Las concentraciones de insulina y glucosa se elevan, y las de glucagón disminuyen.(31) La glucosa que excede las demandas de combustible es almacenada como glucógeno en hígado y músculo o convertida a lípido y almacenada en tejido adiposo.(32) Esta es la única fase durante la cual el hígado es un usuario neto de glucosa y la gluconeogénesis es poco usada.(33)
2. Fase postabsortiva: la insulina retorna a niveles basales, el glucagón se incrementa, y el hígado es llamado a producir glucosa,(34) la cual se deriva principalmente del glucógeno almacenado. El mayor usuario de glucosa durante esta fase es el cerebro, el cual oxida exclusivamente glucosa.(35) Otros consumidores obligados de glucosa como los glóbulos rojos y la médula adrenal son especialmente activos durante este período.(36) Los músculos y el tejido adiposo, sin embargo, usan glucosa a una tasa más baja comparada con la primera fase.(37) El glucógeno presente en el hígado después de una noche de ayuno (90 g en adultos), es suficiente para cubrir los requerimientos de los tejidos periféricos, sólo durante medio día.(38)
3. Fase de ayuno: esta fase comienza inmediatamente después de una noche de ayuno fisiológico.(39) La gluconcongénesis progresivamente reemplaza al glucógeno como mayor fuente de glucosa sanguínea.(40) Los depósitos de glucógeno están agotados y el cerebro no comienza a utilizar todavía cuerpos cetónicos en cantidades significantes, lo que será efectivo en el ayuno avanzado.(41)
ADAPTACIONES METABÓLICAS
Durante el primer y segundo trimestre de la gestación, la hierfagia materna estimula el aumento de peso, el depósito de grasa, y el incremento en índice de masa magra.(42) Además se produce un incremento marcado en los niveles de leptina e insulina séricas.(43) La sensibilidad de los tejidos a la insulina es normal o se encuentra aumentada,(44) y debido al consumo de glucosa por la placenta y al crecimiento fetal, la madre se encuentra predispuesta a la hipoglucemia del ayuno.(45) Durante el tercer trimestre del embarazo, la sensibilidad de los tejidos maternos a la insulina disminuye;(46) la utilización de glucosa por los tejidos maternos es menor, a pesar del aumento marcado de la producción de insulina y de la secreción de insulina estimulada por la glucosa.(47) La resistencia a la insulina promueve entonces la lipólisis y la cetonemia del ayuno, así como la hiperglucemia postprandial, (48) con lo cual una hay una mayor oferta de nutrientes al feto. El transporte placentario de nutrientes estimula la elevación de la insulina fetal, (49) lo que promueve el crecimiento del feto con incremento del acúmulo de tejido graso y el aumento de las reservas de glucógeno hepático.(50)
El desarrollo de la resistencia materna a la insulina coincide con incrementos en las concentraciones séricas de las hormonas lactogénicas prolactina (PRL),(51) lactógeno placentario (PL),(52) y hormona del crecimiento placentario humano (hGH-V).(53) Los lactógenos y los somatógenos reducen la sensibilidad a la insulina en los adipositos y células del músculo esquelético,(54,55) y estimulan la replicación de las células beta, la transcripción del gen de la insulina, y la secreción de insulina dependiente de glucosa en los islotes pancreáticos, (56) todo lo cual es responsible de la resistencia a la insulina y de la hiperinsulina presente en la gestación avanzada.(57-61)
TRANSPORTE PLACENTARIO DE GLUCOSA
La glucosa se encuentra permanentemente a disposición en la circulación materna, cruzando a través de la placenta mediante un sistema de transporte el cual se une a moléculas de glucosa selectivamente,(62) con la limitante de poder ser saturado, como en el caso del transporte facilitado, aunque no a niveles fisiológicos de glucosa materna, (63) y poder tener competencia con otras sustancias. De acuerdo con esto, la transferencia es determinada mediante el gradiente materno-fetal, el flujo sanguíneo en ambos lados, y la morfología de la placenta.(64) Los transportadores de glucosa placentarios son independientes de insulina y por lo tanto, la insulina solo puede alterar la transferencia de glucosa indirectamente causando cambios en los niveles arteriales de glucosa fetal o materna.(65) Así como lo hace con el oxígeno, la placenta toma la cantidad de glucosa que necesita.(66,67)
Debido a que en la unidad feto-placentaria y en el metabolismo energético materno la leptina ha surgido últimamente como un factor metabólico importante, y dado que no fue mencionado en la revisión de lípidos en el embarazo 5 es conveniente actualizarnos incluyendo una sección dedicada a esta hormona en la presente revisión.
LEPTINA
La leptina es una hormona producida predominantemente por las células del tejido adiposo.(68,69) Los niveles circulantes de leptina son proporcionales a la masa de tejido adiposo. Por eso puede ser considerada como una señal del organismo para mostrar su nivel de reservas energéticas.(70) El receptor de la leptina (incluyendo todas las isoformas de mRNA) es expresado en muchos tejidos. Dicha hormona ejerce sus efectos directamente sobre el sistema nervioso central, para la modificación del metabolismo energético, es decir, disminuyendo la ingestión de alimento, incrementando el gasto de energía y disminuyendo la eficiencia metabólica. Varios tejidos, entre ellos el epitelio de la glándula mamaria,(71) la placenta,(72) el fundus gástrico,(73) y músculo (74) pueden producir leptina. Otras funciones relacionadas con el metabolismo óseo, la hematopoyesis y la angiogenésis han sido descritas.(75-77) Además han sido encontrados efectos sobre la madurez sexual, vía receptores o neuronas hipotalámicas que hacen sinapsis directa o indirectamente con neuronas hipotalámicas para la producción de hormona liberadora de gonadotropina.(78) La hormona liberadora de gonadotropina causa secreción pituitaria de la hormona folículo-estimulante y de hormona luteínizante.(79)
El tejido adiposo materno, es la única fuente significante de leptina en la madre, y no la relación feto/placenta como podría pensarse. Los niveles de leptina en el segundo y tercer trimestre del embarazo llegan hasta el 150% a 200% de los niveles encontrados durante el primer trimestre, o en mujeres no gestantes.(80,81) Sin embargo, la placenta es una fuente significativa de la leptina circulante en la madre, y podemos decir entonces que los niveles de leptina materna son la sumatoria de la producida por el tejido adiposo materno y por la placenta. Los niveles de leptina en sangre de cordón están generalmente correlacionados con el peso fetal,(82,83) pero muestran una mejor correlación con la masa grasa del neonato.(84) Las concentraciones más altas de leptina en sangre arterial comparadas con la de sangre venosa, muestran que los niveles de leptina en sangre de cordón reflejan la producción de leptina fetal.(85) Presumiblemente la leptina es producida por tejido adiposo, pero es posible que otros tejidos fetales contribuyan, como lo sugiere el hallazgo de que fetos de madres con diabetes mellitus gestacional presentan niveles de leptina más altos, lo cual ha sido correlacionado con el contenido de grasa abdominal.(86)
Como podemos ver entonces, de acuerdo a las funciones y efectos de esta hormona, los altos niveles de leptina observados en el embarazo, son de alguna manera contrarios a la homeostasis energética durante la gestación.
Cabría esperar que durante la gestación se presentaran bajos niveles de leptina, ya que dicha hormona incrementa la ineficiencia metabólica y disminuye la ingesta de alimentos. Tal parece que el embarazo, (al igual que la obesidad, otra condición caracterizada por niveles altos de leptina), es un estado de resistencia a la leptina, y por eso la hiperleptinemia podría ser una situación compensatoria.(87) Sin embargo, la mayoría de las teorías sobre este tópico en particular son diversas y necesitan todavía ser comprobadas.
Las funciones de leptina en placenta y en el desarrollo fetal, así como otro tipo de funciones en general, son revisadas por diversos autores.(88-90)
DIABETES EN EL EMBARAZO
Durante el embarazo podemos contar con mujeres diabéticas propiamente dichas, es decir, aquellas que la han sufrido antes del embarazo; mujeres que la desarrollan durante el embarazo (diabetes gestacional); y un tercer grupo de mujeres en las cuales la intolerancia a la glucosa es excesiva, pero no francamente diabéticas, conformando el grupo de tolerancia anormal a la glucosa del embarazo.(91) Dentro del primer grupo se encuentran aquellas madres insulino-dependientes y no insulino-dependientes.
Se debe tener en cuenta la tendencia a la hiperglucemia post-prandial fisiológica del embarazo, o estado de tolerancia a la glucosa, pero ante todo, debemos recordar que los factores de riesgo para la presentación de un espectro de intolerancia a la glucosa, el cual puede aparecer posterior a la segunda mitad de la gestación, con implicaciones patológicas, dependen de la edad materna, la historia familiar de diabetes, los factores étnicos, y la obesidad;(92) sin que hasta el momento se cuente con una prueba estándar a nivel internacional para su diagnóstico.(93)
Los controles estrictos de los niveles de glucosa sanguínea, con el objetivo de que sean mantenidos en el ayuno a un valor por debajo de 5 mmol/L y en estado postprandial por debajo de 7 mmol/L han sido valiosos en la reducción de la mortalidad y morbilidad perinatal.(94)
La hiperglucemia materna produce hiperglucemia fetal, generando hiperplasia pancreática del feto, lo que conlleva a hiperinsulinemia fetal con estímulo anormal del crecimiento fetal y consecuencias tales como macrosomía, organomegalia, eritropoyesis incrementada, y disminución de la producción de surfactante, todo ello pudiendo originar a su vez, parto vaginal traumático, cardiomiopatía hipertrófica con hipertrofia septal y hepatomegalia, hipoglucemia neonatal, policiteia neonatal y enfermedad de la membrana hialina respectivamente.(95)
BIBLIOGRAFÍA
1. Illingworth PJ, Jung RT, Howie PW, et al. Reduction in postprandial energy expenditure during pregnancy. British Med J 1987;294:1573. [ Links ]
2. Strayer L. Biochemistry. 4th Edition. New York: W H Freeman and Company; 1995. p. 463-87. [ Links ]
3. Bisdec JT, James WPT. Menstrual cycle hormonal changes and energy expenditure. Proceed Nut Soc 1984; 43: 143A. [ Links ]
4. Lawrence M, Lawrence F, Coward WA, Cole TJ, Whitehead RG. Energy expenditure and energy balance during pregnancy and lactation in The Gambia. En: Nestlé Foundation annual report. Laussane: Nestlé Foundation; 1986. p. 77-80. [ Links ]
5. Osorio JH. Embarazo y metabolismo de las proteínas. Rev Colomb Obstet Ginecol 1999;50:127-32. [ Links ]
6. Osorio JH. Metabolismo de los lípidos durante el embarazo. Rev Colomb Obstet Ginecol 2000; 51:113-7. [ Links ]
7. Campbell-Brown M, Hytten FE. Nutrition. En: Chamberlain G, Broughton F. Eds.Clinical physiology in obstetrics. London: Blackwell Science; 1998. p. 168-70. [ Links ]
8. Ravnikar V, Metzger BE, Freinkel N. Is there a risk of accelerated starvation in normal human pregnancy? Diabetes 1978;27:463. [ Links ]
9. Shambaugh GE, Koelher RA, Yokoo H. Fetal fuels. III. Ketone utilization by fetal heptocyte. Am J Physiol 1978;235:E-330-7. [ Links ]
10. Palacin M, Lasunción MA, Herrera E. Transfer from mother to fetus of L-alanine and glycerol in fed and 48 h-starved pregnant rats. Biochem Soc Trans 1983;11:731-2. [ Links ]
11. Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochem J 1993;295:329-41. [ Links ]
12. Chaves JM, Herrera E. In vitro glycerol metabolism in pregnant rat. Biol Neonate 1980;37:172-9. [ Links ]
13. Knoop RH, Montes A, Warth MR. Carbohydrate and lipid metabolism in normal pregnancy. Food and nutrition board: Laboratory indices of nutritional status in pregnancy. Nat Acad Sci 1978:35-88. [ Links ]
14. Hahn D, Blaschitz A, Korgun ET, et al. From maternal glucose to fetal glycogen: expression of key regulators in the human placenta. Mol Hum Reprod 2000;17:1173-8. [ Links ]
15. Marx J. Unraveling the causes of diabetes. Science 2002;296:686-9. [ Links ]
16. Duro D, Rising R, Cedillo M, et al. Association between infantile colic and carbohydrate malabsorption from fruit juices in infancy. Pediatrics 2002;109:797-805. [ Links ]
17. Misra A, Chaudhary D, Vikram NK, et al. Insulin resistance and clustering of atherogenic risk factors in women belonging to low socio-economic strata in urban slums of North India. Diabetes Res Clin Pract 2002;56:73-5. [ Links ]
18. Drevets WC, Price JL, Bardgett ME, et al. Glucose metabolism in the amygdala in depression: relationship to diagnostic subtype and plasma cortisol levels. Pharmacol Biochem Behavior 2002;71:431-47. [ Links ]
19. Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bel-2 regulated. Diabetes 2002;51:1437-42. [ Links ]
20. Hoyer S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. J Neural Transmission 2002;109:341-60. [ Links ]
21. Grunfeld C. HIV protease inhibitors and glucose metabolism. AIDS 2002;16:925-6. [ Links ]
22. Luke B. Nutritional influences on fetal growth. Clin Obstet Gynecol 1994;37:538-49. [ Links ]
23. Lawson CJ, Homewood J, Taylor AJ. The effects of L-glucose on memory in mice are modulated by peripherally acting cholinergic drugs. Neurobiol Learn Mem 2002;77:17-28. [ Links ]
24. Metzger BE, Phelps RL, Freinkel N, Navickas IA. Effects of gestational diabetes on diurnal profiles of plasma glucose, lipids, and individual amino acids. Diabetes Care 1980;3:402-9. [ Links ]
25. Loebstein R, Koren G. Clinical relevance of therapeutic drug monitoring during pregnancy. Ther Drug Monit 2002;24:15-22. [ Links ]
26. Foster DW. From glycogen to ketones-and back. Diabetes 1984;33:1188-99. [ Links ]
27. Barnea FR. Current progress in early pregnancy investigation and the steps ahead. Early Pregnancy 2000;4:1-4. [ Links ]
28. McGarry JD. Glucose-fatty acid interactions in health and disease. Am J Clin Nutr 1998;67(suppl): 500S-504S. [ Links ]
29. Illingworth PJ, Jung RT, Howie PW, et al. Reduction in postprandial energy expenditure during pregnancy. Br Med J 1987;294:1573. [ Links ]
30. Bisdec JT, James WPT. Menstrual cycle hormonal changes and energy expenditure. Proceed Nut Soc 1984;43:143A. [ Links ]
31. Lawrence M, Lawrence F, Coward WA, Cole TJ, Whitehead RG. Energy expenditure and energy balance during pregnancy and lactation in The Gambia. En: Nestlé Foundation annual report. Laussane: Nestlé Foundation; 1986. p. 90-101. [ Links ]
32. Campbell-Brown M, Hytten FE. Nutrition. In: Chamberlain G, Broughton F, eds. Clinical physiology in obstetrics. London: Blackwell Science; 1998. p. 168-70. [ Links ]
33. Freinkel N. Effects of the conceptus on maternal metabolism during pregnancy. En: Leibel W. On the nature and treatment of diabetes. London; 1965. p. 679-91. [ Links ]
34. Shambaugh GE, Koelher RA, Yokoo H. Fetal fuels. III. Ketone utilization by fetal hepatocyte. Am J Physiol 1978;235:E-330-E337. [ Links ]
35. Palacin M, Lasuncion MA, Herrera E. Transfer from mother to fetus of L-alanine and glycerolin fed and 48 h-starved pregnant rats. Biochem Soc Trans 1983;11:731-2. [ Links ]
36. Zorzano A, Lasuncion MA, Herrera E. Role of the availability of substrates on hepatic and renal gluconeogenesis in the fasted pregnant rat. Metabolism 1986;35:297-303. [ Links ]
37. Chaves JM, Herrera E. In vitro glycerol metabolism in pregnant rat. Biol Neonate 1980;37:172-9. [ Links ]
38. Friedman MI. Fuel partitioning and food intake. Am J Clin Nutr 1998;67(suppl):513S-518S. [ Links ]
39. Iozzo P, Chareonthaitawee P, Terlizzi M, et al. Regional myocardial blood flow and glucose utilization during fasting and physiological hyperinsulinemia in humans. Am J Physiol Endocrinol Metab 2000;282:E1163-71. [ Links ]
40. Knoop RH, Montes A, Warth MR. Carbohydrate and lipid metabolism in normal pregnancy. Food and nutrition board: Laboratory indices of nutritional status in pregnancy. Nat Acad Sci 1978:35-88. [ Links ]
41. Paradisi G, Fulghesu AM, Ferrazzani S, et al. Endocrino-metabolic features in women with polycystic ovary syndrome during pregnancy. Hum Reprod 1998;13:542-6. [ Links ]
42. Caruso A, Ferrazzani S, De Carolis S. Gestational hypertension but not pre-eclampsia is associated with insulin resistance syndrome characteristic. Hum Reprod 1999;14:219-23. [ Links ]
43. Anim-Nyame N, Hills FA, Sooranna SR. A longitudinal study of maternal plasma insulin-like growth factor binding protein-1 concentrations during normal pregnancy and pregnancies complicated by pre-eclampsia. Hum Reprod 2000;15:2215-9. [ Links ]
44. Holmes RP, Holly JMP, Soothill PW. Maternal serum insulin-like growth factor binding protein-2 and -3 and fetal growth. Hum Reprod 1999;14:1879-84. [ Links ]
45. Ogueh O, Miell JP, Jones JC, et al. Antenatal dexamethasone and the growth hormone-insulin-like growth factor axis. Hum Reprod 2000;15:1403-6. [ Links ]
46. Yohichi O, Chizu Y, Toshihiro Y, et al. Hepatocyte growth factor concentration in the early second-trimester amniotic-fluid does not predict fetal growth at birth. Hum Reprod 1999;14:2625-8. [ Links ]
47. Langford K, Nicolaides K, Miell JP. Maternal and fetal insulin-like growth factors and their binding proteins in the second and third trimesters of human pregnancy. Hum Reprod 1998;13:1389-93. [ Links ]
48. Vannini P. Pregnancy and diabetes: physiopathological aspects. Minerva Endocrinologica 1994;19: 45-50. [ Links ]
49. Hoiriis Nielsen J, Nielsen V, Molsted Pedersen L, et al. Effects of pregnancy hormones on pancreatic islets in organ culture. Acta Endocrinol 1986;111:336-41. [ Links ]
50. Freemark M. (2001) Ontogenesis of prolactin receptors in the human fetus: roles in fetal development. Biochem Soc Trans 2001;29:38-41. [ Links ]
51. Irving RJ, Walker BR, Noon JP, et al. Microvascular correlates of blood pressure, plasma glucose, and insulin resistance in health. Cardiovascular Res 2002;53:271-6. [ Links ]
52. Stevens-Simon C, Thureen P, Barrett J, et al. Regional body fat distribution and insulin resistance during adolescent pregnancy. J Am Diet Assoc 2002;102:563-5. [ Links ]
53. Baron AD. Insulin resistance and vascular function. J Diabetes Complications 2002;16:92-102. [ Links ]
54. Ryan EA, Enns L. Role of gestational hormones in the induction of insulin resistance. J Clin Endocrinol Metab 1998;67:341-7. [ Links ]
55. Leturque A, Hauguel S, Sutter Dub MT, Maulard P, Girard J. Effects of placental lactogen and progesterone in insulin stimulated glucose metabolism in rat muscles in vitro. Diabete Metab 1989;15:176-81 [ Links ].
56. Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, Friesen HG, Sorenson RL. Effect of homologous placental lactogens, prolactina, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of the islet function during pregnancy. Endocrinology 1993;132:879-87. [ Links ]
57. Brelje TC, Sorenson RL. Role of prolactin versus growth hormone on islet B-cell proliferation in vitro: implications for pregnancy. Endocrinology 1991;128:45-57. [ Links ]
58. Sorenson RL, Brelje TC, Roth C. Effects of steroid and lactogenic hormone on islets of Langerhans: a new hypothesis for the role of pregnancy steroids in the adaptation of islets to pregnancy. Endocrinology 1993;133:2227-34. [ Links ]
59. Brelje TC, Scharp DW, Lacy PE, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993;132:879-87. [ Links ]
60. Petryk A, Fleenor D, Driscoll P, Freemark M. Prolactin induction of insulin gene expression: the roles of glucose transporter-2. Journal of Endocrinology 2000;164:277-86. [ Links ]
61. Fleenor D, Petryk A, Driscoll P, Freemark M. Constitutive expression of placental lactogen in pancreatic beta cells: effects on cell morphology, growth, and gene expression. Pediatr Res 2000;47:136-42. [ Links ]
62. Haggarty P, Allstaff S, Hoad G, et al. Placental nutrient transfer capacity and fetal growth. Placenta 2002;23:86-92. [ Links ]
63. Osmond DT, King RG, Brennecke SP, et al. Placental glucose transport and utilization is altered at term in insulin-treated, gestational-diabetic patients. Diabetologia 2002;44:1133-9. [ Links ]
64. Stephenson T, Symonds ME. Maternal nutrition as a determinant of birth weight. Arch Dis Child Fetal Neonatal Ed 2002;86: F4-6. [ Links ]
65. Harding J. Placental Physiology. En: Gluckman P, Heymann M. Eds. Perinatal and pediatric pathophysiology: A clinical perspective. London: Edward Arnold; 1993. [ Links ]
66. Lachili B, Hininger I, Faure H, et al. Increased lipid peroxidation in pregnant women after iron and vitamin C supplementation. Biol Trace Elem Res 2001;83:103-10. [ Links ]
67. Battaglia FC, Meschia G. Fetal nutrition. Annu Rev Nutr 1988;8:43-61. [ Links ]
68. Tsumanuma I, Jin L, Zhang S, et al. Leptin signal transduction in the HP75 human pituitary cell line. Pituitary 2000;3: 211-20. [ Links ]
69. Baba T, Kanda T, Yoshida A, et al. Reciprocal changes in leptin and tumor necrosis factor-alpha with exercise in insulin resistant rats. Res Commun Mol Pathol Pharmacol 2000;108:133-43. [ Links ]
70. Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature 1996;382:250-2. [ Links ]
71. Smith-Kirwin SM, O'Connor DM, De Johnston J, et al. Leptin expression in human mammary epithelial cells. J Clin Endocrinol Metab 1998;83:1810-3. [ Links ]
72. Linnemann K, Malek A, Shneider H, Fush C. Physiological and pathological regulation of feto/placento/maternal leptin expression. Biochem Soc Trans 2001;29:86-90. [ Links ]
73. Bado A, Levasseur S, Attoub S, et al. The stomach is a source of leptin. Nature 1998;394:790-3. [ Links ]
74.Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 1998;393:684-8. [ Links ]
75. Ducy P, Amling M, Takeda S, et al. Leptin inhibes bone formation through a hypothalamic relay: a central control of bone mass. Cell 2000;100:197-207. [ Links ]
76. Gainsford T, Willson TA, Metcalf D, et al. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc Nat Acad Sci USA 1996;93:14564-8. [ Links ]
77. Sierra-Honigmann MR, Nath AK, et al. Biological action of leptin as an angiogenic factor. Science 1998;281:1683-6. [ Links ]
78. Yu WH, Kimura M, Walczewska A, Karanth S, McCann SM. Role of leptin in hypothalamic-pituitary function. Proc Nat Acad Sci USA 1997;94:1023-8. [ Links ]
79. Iwashita S, Tamida N, Tarui N, et al. Direct measurement of renal sympathetic nervous activity in high-fat diet-related hypertensive rats. Life Sci 2002;71:537-46. [ Links ]
80. Masuzaki H, Ogawa Y, Sagawa N, et al. Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med 1997;3:1029-33. [ Links ]
81. Butte NF, Hopkinson JM, Ellis KJ, et al. Changes in fat-free mass and fat mass in postpartum women: a comparison of body composition models. Int J Obes Relat Metab Disord 1997;21:874-80. [ Links ]
82. Schubring C, Kiess W, Englaro P, et al. Leptin concentrations in amniotic fluid, venous and arterial cord blood and maternal serum: high leptin synthesis in the fetus and inverse correlation with placental weight. Eur J Pediatr 1996;155:830. [ Links ]
83. Schekhawat PS, Garland JS, Shivpuri C, et al. Neonatal cord blood leptin: its relationship to birth weight, body mass index, maternal diabetes, and steroids. Pediatr Res 1998;43:338-43. [ Links ]
84. Clapp JF 3rd, Kiess W. Cord blood leptin reflects fetal fat mass. J Soc Gynecol Investig 1998;5:300-3. [ Links ]
85. Ong K,Kratzsch J, Kiess W, et al. The ALSPAC Study Team. Circulating IGF-I levels in childhood are related to both current body composition and early postnatal growth rate. J Clin Endocrinol Metab 2002;87:1041-4. [ Links ]
86. Cetin I, Morpurgo PS, Radaelli T, et al. Fetal plasma leptin concentrations: relationship with different intrauterine growth patterns from 19 weeks to term. Pediatr Res 2000;48:646-51. [ Links ]
87. Orbak Z, Darcan S, Coker M, et al. Maternal and fetal serum insulin-like growth factor-I (IGF-I) IGF binding protein-e (IGFBP-3), leptin levels and early postnatal growth in infants born asymmetrically small for gestational age. J Pediatr Endocrinol Metab 2001;14:1119-27. [ Links ]
88. Harris RB. Leptin--much more than a satiety signal. Annu Rev Nutr 2000;20:45-75. [ Links ]
89. Reitman ML, Bi S, Marcus-Samuel B, et al. Leptin and its role in pregnancy and fetal development- an overview. Biochem Soc Trans 2001;29:68-72. [ Links ]
90. Hoggard N, Haggarty L, Thomas L, Lea RG. Leptin expression in placental and fetal tissues: does leptin have a functional role? Biochem Soc Trans 2001;29:57-63. [ Links ]
91. Lucas MJ. Diabetes complicating pregnancy. Obstet Gynecol Clin North Am 2001;28:513-6. [ Links ]
92. Landon MB. Diabetes mellitus and other endocrine diseases. En: Gabbe SG, Niebyl JR, Simpson JL. Obstetrics. New York: Churchill Livingstone; 1991. p. 112. [ Links ]
93. Greene MF. Prevention and diagnosis of congenital abnormalities in diabetic pregnancies. Clin Perinatol 1993;20:533-47. [ Links ]
94. Pedersen JF. The pregnant diabetic and her newborn. Baltimore: Williams and Wilkins; 1967. p. 126. [ Links ]
95. Haugel-de Mouzon S, Shafrir E. Carbohydrate and fat metabolism and related hormonal regulation in normal and diabetic placenta. Placenta 2001;22:619-27. [ Links ]


{kind=link}