










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
Print version ISSN 0034-7434On-line version ISSN 2463-0225
Rev Colomb Obstet Ginecol vol.57 no.2 Bogotá June 2006
* Especialista en Genética Médica. Instituto de Genética Humana. Pontificia Universidad Javeriana. Bogotá, Colombia.
** Director Científico. Medifertil. Programa de Medicina Reproductiva. Bogotá, Colombia. Correo electrónico: jimadero@cable.net.co
*** Especialista en Ginecología y Obstetricia y Medicina Materno Fetal. Facultad de Medicina. Universidad del Rosario. Bogotá, Colombia.
+ Medifertil. Programa de Medicina Reproductiva.
RESUMEN
El Síndrome de Turner (ST) es la patología más frecuente que compromete los cromosomas sexuales, es causada por la ausencia completa o parcial del cromosoma X. Las implicaciones reproductivas de estos pacientes que se constituyen en infertilidad por una falla ovárica prematura y disgenesia gonadal, sugieren que el manejo indicado es la donación de óvulos asociada con la generación de embriones in vitro y su posterior transferencia, previa preparación endometrial. En este artículo se informan dos casos de ST manejados con ovodonación y sus implicaciones clínicas en el embarazo: Desproporción cefalo-pélvica por talla baja, complicaciones cardiovasculares que generan linfedema, lesiones aórticas y preeclampsia.
Palabras clave: Síndrome de Turner, infertilidad, Donación de ovocito, Complicaciones del embarazo.
SUMMARY
Turner’s syndrome (TS) is the most frequent pathology compromising the sexual chromosomes, being caused by the complete or partial absence of chromosome X. The reproductive implications for these patients (i.e. infertility caused by early ovarian failure and gonad disgenesis) have suggested that the indicated management for this condition lies in anonymous ovodonation, followed by embryos being produced in vitro and their subsequent transfer, after suitable endometrial preparation. This article reports oocyte donation to two patients suffering from TS and their clinical implications for pregnancy: cephalo-pelvic imbalance due to small maternal size and cardiovascular complications producing lymphoedema, aortic dissection and preeclampsy.
Key words: Turner syndrome, infertility, oocyte donation, fetal-maternal complications.
INTRODUCCIÓN
El Síndrome de Turner (ST), descrito por Otto Ullrich en 1930 y Henry Turner en 19381 es la alteración más frecuente que compromete los cromosomas sexuales, siendo causada por ausencia completa o parcial del cromosoma X y en ocasiones acompañado por mosaicismos.2 Afecta 1 de 2.000 a 5.000 recién nacidos de sexo femenino3 y se estima que el 3% de las concepciones de embriones y fetos femeninos tienen esta patología, llegándose a abortar espontáneamente en el primer trimestre entre el 95 al 99%.4 Con respecto a las pérdidas detectables, del 10 al 15% son por fetos con ST. En el 50% de los casos hay ausencia de todo el cromosoma X, observándose un complemento cromosómico 45, X0; el otro 50% de los casos presentan múltiples anomalías cromosómicas como mosaicismos, deleciones parciales o translocaciones. Las anomalías tipo deleción pueden ser de brazo largo de cromosoma X, de brazo corto, cromosomas X en anillo, translocaciones autosómicas o rearreglos complejos.5
Dentro de los mosaicismos más comunes se encuentran 45,X/46,XX;45,X/46,XXiq y 45,X/46,XY6 siendo el grado de mosaicismo en ocasiones no relacionado con la severidad del cuadro ni con componentes parentales, excepto en el último caso donde la presencia del cromosoma Y predispone a la aparición de gonadoblastoma7 hasta en un 30%.8 En varias ocasiones no es sospechado clínicamente el ST mosaico hasta la adolescencia, existiendo ca-sos de embarazos naturales con este complemento cromosómico,9,10 de los cuales la mayoría terminan en abortos o son mujeres con cuadros de aborto recurrente debido a defectos genéticos fetales, alteraciones de la vasculatura útero ovárica con sub perfusión e hipoplasia uterina y anormalidades uterinas subclínicas.11
Esta patología se caracteriza por la presencia de la triada: talla baja, falla ovárica y otras anomalías como cuello alado y/o corto y cúbitus valgus, la cual no es completamente evidente desde el nacimiento, como si pueden serlo otros signos (linfedema, cardiopatías, micrognatia, tórax amplio, teletelia, facies característica, entre otros).12 Su mortalidad es tres veces mayor con respecto mujeres 46, XX por el compromiso multiorgánico que conlleva (cardiovascular, endocrino, esquelético, sensorial)13 y por ende su expectativa de vida es 13 años menor primordial-mente por cardiopatías y ateroesclerosis.14
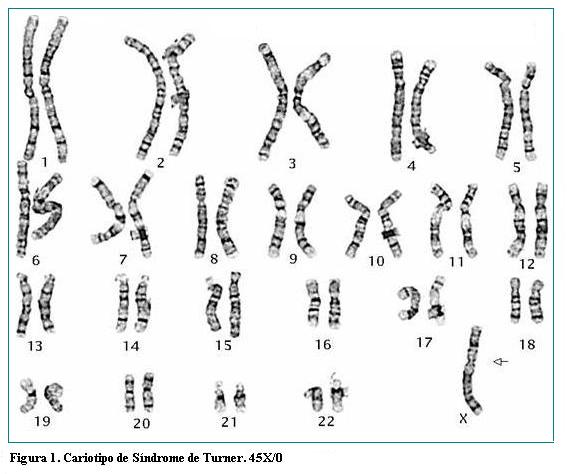
Es una enfermedad que se puede diagnosticar pre y posnatalmente, siendo este último el periodo más frecuente del diagnóstico; el 20% de los casos se diagnostica en la niñez y la mayoría son diagnosticados en la adolescencia cuando se evidencia la baja talla, retraso de la pubertad o amenorrea primaria. Este diagnóstico puede ser retrasado aún más a etapa adulta (entre el 10% de los casos) cuando la mujer presenta amenorrea primaria, secundaria o infertilidad. Luego del nacimiento, los pilares diagnósticos son la exploración clínica detallada en búsqueda de los signos más comunes de esta patología como la baja talla, la falla ovárica y los signos menores y mayores acompañantes y el soporte paraclínico como ecocardiogramas, radiografías, niveles hormonales en sangre, ecografías renales y la confirmación diagnóstica por cariotipo (Figura 1). En este reporte de casos, el diagnóstico de ingreso de las pacientes al programa de infertilidad de esta institución fue infertilidad secundaria por falla ovárica prematura y baja talla. Posteriormente el estudio más detallado tanto clínico, operatorio y paraclínico de la paciente hizo virar el enfoque, diagnóstico y manejo.
CASOS CLÍNICOS
Paciente 1
Paciente de 37 años quien consulta a Medifertil por infertilidad primaria, la menarquia fue a los 13 años, con ciclos de 22-25 x 3 días y menopausia a los 32 años. G0P0. Adicionalmente presentaba depresión manejada con litio el cual suspendió un mes antes de la consulta; se le había realizado un laparoscopia diagnóstica con reporte desconocido. Tiene un hermano con azoospermia y una hermana con falla ovárica prematura.
Al examen físico: baja talla simétrica (145 cm), peso 62,5 kg. Implantación normal de cabello y orejas, prognatismo, cuello normal, senos voluminosos, ausencia de teletelia, examen cardiopulmonar sin alteraciones, cubitus valgus bilateral, braquidactilia bilateral en manos y pies y pulgares prominentes en 4 extremidades, genitales externos de nulípara. Se evalúan útero y ovarios por ecografía apreciando útero disminuido de volumen, endometrio lineal, atrófico. No se observaron los ovarios. Histeroscopía con cavidad uterina disminuida de volumen.
Se solicita cariotipo bandeo G por sospecha de disgenesia gonadal y niveles de FSH y Estradiol para estudio de falla ovárica prematura, así como espermograma como estudio de factor masculino. Resultados de niveles de FSH 8,1 mIU/ml y estradiol menor de 20 pgr/ml. El espermograma inicial mostró un número de 120 millones por ml, un volumen de 1,1 ml, motilidad tipo A (rápida) del 14%, B (lenta) del 35%, C del 15% y D del 36%. El 14% de los espermatozoides eran normales. Bacterias +. Se inicia manejo con ciprofloxacina, el espermograma post tratamiento mostró una mejoría en la motilidad tipo A la cual aumentó del 14 al 19%, la motilidad tipo B alcanzó el 41%, sin bacterias, con vitalidad del 77% y formas normales del 20%. El cariotipo de la paciente reportó 3% de metafases con complemento cromosómico anormal de 45, X0; 97% complemento cromosómico normal de 46,XX. Mosaico 46,XX/45,X0. Se realizó tratamiento con valerianato de estradiol en dosis ascendentes hasta 6 mg logrando endometrio de 9 mm y recuperación del volumen uterino, luego progesterona por 11 días; con base en el resultado anterior se inició esquema de preparación endometrial transfiriéndose cuatro embriones producto de donación de óvulos. Conviene aclarar que actualmente solo transferimos dos embriones. Diez días después se determinaron niveles de Beta hCG: 27,8 mUI/ml. Se realiza seguimiento ecográfico con la presencia de dos sacos intrauterinos con vesícula vitelina y embriones con actividad fibrilar; el embarazo transcurre sin complicaciones hasta la semana 30 cuando consulta a su médico por cuadro de cefalea y fosfenos, se le detecta una elevación de la tensión arterial, debiendo iniciar estudio por cuadro de preeclampsia. En la semana 32 sele inicia manejo a base de hipotensores y útero inhibidores por amenaza de parto prematuro debiéndose hospitalizar en la semana 35. En la semana 37 se le realizó una cesárea por cuadro de preeclampsia, se obtuvieron dos recién nacidos sanos: femenino de 2.470 gr y talla de 48 cm y masculino de 2.570 gr y talla de 49 cm, ambos con Ballard de 37 semanas y Apgar normal. Se concluye que la paciente tuvo una infertilidad primaria, secundaria a una falla ovárica prematura debida a una disgenesia gonadal por un Síndrome de Turner variedad mosaico, en donde el manejo indicado es la donación de óvulos.
Paciente 2
Paciente de 34 años quien consulta a Medifertil por cuadro de infertilidad primaria secundario a disgenesia gonadal diagnosticada por amenorrea primaria y por laparoscopia que mostró un útero moderadamente hipoplásico con bandas fibrosas en sitio de los ovarios (la patología reporta “segmento de tejido fibroso sin alteraciones, no se reconoce tejido ovárico”). En 1989 se le realizó una laparotomía para biopsia de ovario (no esta el resultado) pero llama la atención que en la descripción quirúrgica dice: ovarios pequeños con folículos. Le formulan manejo a base de estrógenos y progestágenos cíclicos el cual suspendió hace 3 años.
Se realizó diagnóstico de disgenesia gonadal con cariotipo 46 XX, el cual se repite en marzo del 2006 reportándose un mosaico 45X/46XX en bandeo GTG. Al examen físico: talla baja proporcionada, cuello corto, cubitus valgus. Cabello de implantación adecuada, teletelia, mamas con Tanner 3. Se realizó ecografía observando útero pequeño con línea endometrial lineal. No hay imágenes de ovarios. Empieza preparación endometrialpara ver respuesta logrando endometrio de 9 mm, en ese momento se le realizó una histeroscopía observando una cavidad uterina aún disminuida de volumen. En ciclo siguiente se le realizó ovodonación, transfiriéndose 4 embriones de buena calidad. Se determinan los niveles de Beta hCG el día 10 post transferencia hallándolos en 50 mUI y el 18 en 2.600 mUI. La ecografía 20 días post transferencia de embriones mostró un saco gestacional único con una vesícula vitelina en su interior y un adecuado halo de implantación. Es hospitalizada a las 30 semanas de gestación por preeclampsia severa, llegando hasta la semana 38 cuando se le realiza cesárea obteniéndose un RN masculino, de 3.133 gr y talla de 51 cm. Se concluye que la paciente tuvo infertilidad por una falla ovárica prematura, secundaria a disgenesia gonadal por Síndrome de Turner, en donde el manejo indicado es la donación anónima de óvulos asociado a la generación de embriones in vitro y su posterior transferencia, previa preparación endometrial.
PERSPECTIVAS GINECO - OBSTÉTRICAS EN EL ST
Desde el punto de vista ginecológico y obstétrico, el manejo del ST posee tres puntos de discusión muy interesantes: opción de fertilidad y reemplazo estrogénico por la disgenesia ovárica, el tratamiento reproductivo y el seguimiento al embarazo como la pelvis pequeña por su talla baja y por último los problemas cardiovasculares que generan linfedema y lesiones aórticas en ST,15 por lo tanto el manejo de esta patología desde el periodo neonatal hasta el periodo adulto requiere del manejo multidisciplinario que comprometa a neonatólogos, pediatras, genetistas, ginecólogos, cardiólogos, entre otros.
Disgenesia ovárica
El desarrollo gonadal embriológico de las pacientes con ST es normal hasta la semana 18 cuando empieza a producirse la depleción de los ovocitos los cuales se hallan en la fase temprana de la profase de la meiosis y continúa hasta los primeros meses y años de vida, dando como consecuencia una estría ovárica compuesta de tejido estromal blanco fibroso el cual no contiene ovocitos o restos foliculares. Sin embargo en los casos de cariotipos con mosaicos hay una escasa cantidad de folículos evidenciado por la presencia de menarquia espontánea hasta en el 12% de las pacientes así como por la evidencia escasa pero presente de embarazos espontáneos en porcentajes del 2 al 7% en pacientes con ST.16-18 Un tercio de los productos de estas gestaciones son abortos o muertes perinatales; el otro tercio presenta recién nacidos con anomalías cromosómicas como Síndrome de Down, ST y malformaciones mayores.
Por otro lado, debido al perfil de hipogonadismo hipergonadotrópico en niñas con ST asociados a la talla baja, se ha discutido ampliamente el momento adecuado para iniciar la administración exógena de estrógenos intentando lograr una talla aceptable aunque sí hay un acuerdo en la administración de hormona del crecimiento a temprana edad. Es así como la inducción farmacológica de la menarquia ha sido pospuesta hasta después de los 14 años con el fin de lograr mayor crecimiento de los núcleos de osificación.
El reemplazo hormonal en mujeres con ST se debe individualizar a cada paciente y a su comorbilidad. Se estima que la administración de estrógenos exógenos por medio de píldoras anticonceptivas no es el esquema óptimo para el manejo de ésta entidad, ya que al requerir una semana al mes de placebo, se tienen 3 meses al año sin reemplazo hormonal; mientras que al administrar estrógenos con esquemas de terapia de suplencia hormonal se logran mejores niveles de estrógenos y volúmenesuterinos mayores sin generar un riesgo aumentado de incidencia de cáncer de seno usando este tipo de estrógenos exógenos.19, 20
Tratamientos de reproducción y embarazos por técnicas de fertilización in vitro en pacientes con ST
Los embarazos espontáneos ocurren solamente entre un 2% a 7% de las pacientes con ST y estos embarazos están asociados a alto riesgo de pérdidas, mortinatos, malformaciones y anomalías cromosómicas.21 La tasa de cesáreas es alta debido a varias condiciones obstétricas propias del ST como la talla baja, que se traduce en pelvis estrecha y por lo tanto desproporción céfalo pélvica y falta de descenso de la presentación cuando hay trabajo de parto, además toda paciente que ingrese a un programa de reproducción asistida debe tener una valoración por cardiología y una ecocardiografía para evaluar previamente problemas aórticos y valvulares.
Se han utilizado de forma exitosa algunas técnicas de reproducción asistida para el manejo de la esterilidad en ST.17Varios protocolos se han descrito para la preparación del endometrio antes de la transferencia de ovocitos donados y fertilizados usando estrógenos y bajas dosis de progestágenos asociado a seguimiento ecográfico diario.18 Las tasas de embarazos de pacientes con ST son similares a las observadas en pacientes sin ST en programas de ovo donación.19, 20 Se han reportado en la literatura tasas de éxito hasta del 40% por ciclo y se logran 50% de nacidos vivos.21 Sin embargo, en otros estudios estas tasas han sido mas bajas posiblemente por algún factor endometrial que interfiere en el adecuado proceso de implantación, siendo mas frecuente las pérdidas en ST universal (45, X0) que en mosaicos.22
Riesgos cardiovasculares
Entre el 23 y el 40% de personas con Síndrome de Turner tienen algún tipo de malformaciones cardiacas congénitas, siendo más común en cariotipos 45, X0 y menos común en Turner por isocromosomas.23, 24 Las complicaciones cardiovasculares ocurren entre el 10 al 44% y son la principal causa de mortalidad asociadas con ST25 siendo las lesiones de corazón izquierdo y aórticas las más importantes. La malformación más frecuente es la válvula aórtica bicúspide la cual puede asociarse o no a coartación aórtica. También son frecuentes la dilatación aórtica y las anomalías del corazón izquierdo, que predisponen a endocarditis infecciosa, requiriendo manejo profiláctico antibiótico antes de ser sometidas a cualquier procedimiento quirúrgico o dental. La dilatación aórtica y la disección presentan especial atención por su frecuencia en ST (8-28%)26 y el riesgo de muerte súbita además de muerte durante el embarazo y posparto.27, 28 Se ha reportado la complicación de dilatación aórtica en el embarazo por lo menos en 81 casos en todo el mundo29 y 4 muertes de pacientes con ST por disección aórtica en embarazos generados por donación de ovocitos,30 proporcionando de forma preliminar una tasa de mortalidad materna en ST del 2%.31 Gracias a esto, se requiere una estrecha evaluación ecocardiográfica y con RNM de corazón. Finalmente, las pacientes con ST tienen un riesgo 3 veces mayor de hipertensión arterial que las pacientes sanas y sólo un 20% se ha probado que sea secundario a causas renales o cardiacas, por lo cual se aconseja siempre una evaluación previa por cardiología para realizar un ecocardiograma y una resonancia en estos casos.
CONCLUSIONES
Debido a los avances en el entendimiento genético y molecular de la fisiopatología del ST, se ha podido mejorar la expectativa y la calidad de vida de estas pacientes desde distintas ramas de la medicina, proporcionándoles también la posibilidad de concebir mediante técnicas de reproducción asistida como la ovodonación con tasas de éxito de embarazo iguales o cercanas a mujeres sin ST sometidas a los mismos procedimientos. Estas mujeres deben ser evaluadas y controladas por un grupo multidisciplinario de forma estricta integrado por los gineco-obstetras, perinatólogos, cardiólogos, internistas y genetistas entre otros, debido a las posibles complicaciones inherentes al ST durante el embarazo como pérdidas fetales, hipertensión, desproporción cefalopélvica y primordialmente problemas cardiovasculares como la dilatación y disección de aorta, con tasas de mortalidad cercanas al 2%.
REFERENCIAS
1. Turner H. A syndrome with infantilism, congenital webbed neck and cubitus valgus. Endocrinology 1938;23:566-74. [ Links ]
2. Ranke MB, Saenger P. Turner’s syndrome. Lancet 2001;358:309-14. [ Links ]
3. Lippe B. Turner syndrome. Endocrinol Metab Clin North Am 1991;20:121-52. [ Links ]
4. Hsu L. Prenatal diagnosis of chromosomal abnormalities through amniocentesis. En: Milunsky A (Ed). Genetic disorder and the Fetus. Baltimore: John Hopkins University Press; 1998. p. 179-248. [ Links ]
5. Turner C, Dennis NR, Skuse NH, Jacobs PA. Seven ring (X) chromosomes lacking XIST locus, six with an unexpectedly mild phenotype. Hum Genet 2000;106:93-100. [ Links ]
6. Grayholt CH, Juul S, Naeraa RW, Hansen J. Prenatal and postnatal prevalence of Turner’s syndrome: a registry study. BMJ 1996:321:16-21. [ Links ]
7. Mathur A, Stekol L, Schatz D, MacLaren NK, Scott ML, Lippe B. The parental origin of the single X chromosome in Turner’s syndrome: lack of correlation with parental age or clinical phenotype. Am J Hum Genet 1991;48:682-6. [ Links ]
8. Verp MS, Simpson JL. Abnormal sexual differentiation and neoplasia. Cancer Genetic Cytogenet 1987;25:191-218. [ Links ]
9. Tarani L, Lampariello G, Raguso G, Colloridi F, Pucarelli J, Pasquino AM, et al. Pregnancy in patients with Turner’s syndrome: six new cases and review of the literature. Gynecol Endocrinol 1998;12:83-7. [ Links ]
10. Check JH, Schubert B, Chase JS. Fetal outcome of triplets in a Turner mosaic. J Perinat Med 1993;21:279-83. [ Links ]
11. Rizk DE, Deb P. A spontaneous and uneventful pregnancy in a Turner mosaic with previous recurrent miscarriages. J Pediatr Adolesc Gynecol 2003;16:87-8. [ Links ]
12. Grayholt CH. Medical problems of adult Turner´s syndrome. Horm Res 2001;56 Suppl 1:44-50. [ Links ]
13. Grayholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in Turner syndrome. J Clin Epidemiol 1998;51:147-58. [ Links ]
14. Price WH, Clayton JF, Collyer S, De Mey R, Wilson J. Mortality ratios, life expectancy, and causes of death in patients with Turner’s syndrome. J Epidemiol Community Health 1986;40:97-102. [ Links ]
15. Conway GS. Considerations for transition from paediatric to adult endocrinology: women with Turner’s syndrome. Growth Horm Res 2004;14Suppl A:S77-84. [ Links ]
16. Kaneko, S. Kawagoe, M. Hiroi, Turner’s syndrome -- review of the literature with reference to a successful pregnancy outcome. Gynecol Obstet Invest 1990;29:81-7. [ Links ]
17. Lutgen P, Trounson A, Leeton J, Findlay J, Wood C, Renou P. The establishment and maintenance and pregnancy using in vitro fertilization and embryo donation in patient with primary ovary failure. Nature 1984;307:174-5. [ Links ]
18. Abir R, Fish B, Nahum R, Orvieto R, Nitke S, Ben Rafael Z. Turner’s syndrome and fertility: current status and possible putative prospect. Hum Reprod Update 2001;7: 603-10. [ Links ]
19. Saenger P, Wikland KA, Conway GS, Davenport M, Grayholt TH, Hintz R, et al. Recommendations for the diagnosis and management of Turner syndrome. J Clin Endocrinol Metab 2001;86:3061-9. [ Links ]
20. Press F, Shapiro HM, Cowell CA, Oliver GD. Outcome of ovum donation in Turner’s syndrome patients. Fertil Steril 1995;64:995-8. [ Links ]
21. Foudila T, Soderstrom-Anttila V, Hovatta O. Turner’s syndrome and pregnancies after oocyte donation. Hum Reprod 1999;14:532-5. [ Links ]
22. Yaron Y, Ochshorn Y, Amit A, Yovel I, Kogosowki A, Lesing JB. Patients with Turner’s syndrome may have an inherent endometrial abnormality affecting receptivity in oocyte donation. Fertil Steril 1996;65:1249-52. [ Links ]
23. Sybert VP. Cardiovascular malformations and complications in Turner syndrome, Pediatrics 1998;101:E11. [ Links ]
24. Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS). J Pediatr 1998;133:688-92. [ Links ]
25. Birdsall M, Kennedy S. The risk of aortic dissection in women with Turner syndrome. Hum Reprod 1996;15:1587. [ Links ]
26. Allen DB, Hendricks SA, Levy JM. Aortic dilation in Turner syndrome. J Pediatr 1986;109:302-5. [ Links ]
27. Nagel TC, Tesch LG. ART and high risk patients! Fertil Steril 1997;68:748-9. [ Links ]
28. Garvey P, Elovitz M, Landsberger E. Aortic dissection and myocardial infarction in a pregnant patient with Turner syndrome. Obstet Gynecol 1998;91(5 pt 2):864. [ Links ]
29. Lin AE, Lippe B, Rosenfeld RG. Further delineation of aortic dilation, dissection and rupture in patients with Turner syndrome. Pediatrics 1998;102:e12. [ Links ]
30. Beauchesne LM, Connolly HM, Ammash NM, Warnes CA. Coartation of the aorta: outcome of pregnancy. J Am Col Cardiol 2001;38:1728-33. [ Links ]
31. Karnis MF, Zimon AE, Lalwani SI, Timmreck LS, Klipstein S, Reindollar RH. Risk of death in pregnancy achieved through oocyte donation in patients with Turner syndrome: a national survey. Fertil Steril 2003;80:498-501. [ Links ]

