










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
Print version ISSN 0034-7434On-line version ISSN 2463-0225
Rev Colomb Obstet Ginecol vol.57 no.4 Bogotá Dec. 2006
* Profesor de obstetricia y ginecología, Departamento de Obstetricia y Ginecología, Hospital Universitario San Ignacio. Facultad de Medicina. Pontificia Universidad Javeriana, Bogotá, Colombia. Fellow de Ginecología Pediátrica y Adolescente, Centro de Medicina Reproductiva y Desarrollo Integral del Adolescente (CEMERA), Facultad de Medicina, Universidad de Chile, Santiago, Chile. Correo electrónico: g.barbosa@javeriana.edu.co.
** Profesor de cirugía y urología pediátrica, Jefe Unidad de Patología del Polo Caudal, Hospital Luis Calvo Mackenna, Facultad de Medicina, Universidad de Chile, Santiago, Chile. Correo electrónico: marioantoniovarela@gmail.com.
RESUMEN
Se presenta el caso clásico de una adolescente con diagnóstico incidental y tardío de síndrome de Mayer-von Rokitansky-Küster-Hauser y lo que consideramos aspectos fundamentales a tener en cuenta para favorecer el diagnóstico precoz y manejo de estas pacientes desde el diagnóstico inicial. Incluimos una descripción detallada de la técnica quirúrgica utilizada para reparar la anomalía, que consistió en una neovaginoplastia sigmoidea en tres tiempos sucesivos por la técnica de Peña modificada. Recalcamos la importancia del manejo multidisciplinario que involucra tanto un equipo médico calificado, como paramédicos y profesionales del área psico-social y de igual forma involucrar en todos los aspectos, la opinión de la paciente y sus padres o tutores.
Palabras clave: agenesia de vagina, ginecología, pediatría.
SUMMARY
The classical case of an adolescent having had incidental and late diagnosis of Mayer-von Rokitansky-Küster-Hauser syndrome is presented, as are aspects considered fundamental in supporting early diagnosis and treatment of these patients from initial evaluation to detailed description of the surgical procedure. This consisted of a sigmoid neovaginoplasty on three consecutive occasions according to modified Pena procedure. The importance of a multidisciplinary approach is stressed, involving a team qualified in medical, paramedical and psycho-social disciplines, as well as involving the patient and parents in all aspects of the treatment.
Keywords: agenesis vagina, gynecology, pediatrics.
INTRODUCCIÓN
El Síndrome de Mayer-von Rokitansky-Küster-Hauser (MRKH) tiene una incidencia de ~ 1:5.000 nacidas vivas. Se caracteriza por agenesia de útero y vagina, pero puede variar desde la total ausencia de estas dos estructuras, hasta la presencia de remanentes uterinos (müllerianos) con o sin tejido endometrial; y del tercio inferior de la vagina, que puede consistir en simplemente una pequeña depresión entre los labios, o tener una longitud de hasta 5 a 6 cm.1 Las mujeres que padecen este síndrome tienen un cariotipo 46XX y un fenotipo femenino normal con desarrollo espontáneo de características sexuales secundarias, dado que el tejido ovárico se desarrolla y funciona normalmente. El tratamiento hormonal es innecesario ya que la producción de estrógenos es normal y de hecho presentan ciclos ovulatorios, así que la reproducción asistida, mediante estimulación ovárica, aspiración de ovocitos y útero subrogado es factible.2,3
Presentamos el caso clásico de una adolescente con diagnóstico incidental de MRKH y lo que consideramos aspectos fundamentales a tener en cuenta para favorecer el diagnóstico precoz y manejo de estas pacientes.
CASO CLÍNICO
CA es una paciente de 15 años y 3 meses de edad quien consultó por dolor abdominal al servicio de Urgencias Pediátricas del Hospital Luis Calvo Mackenna, un hospital público universitario de alto nivel de complejidad en Santiago de Chile.
En ecografía abdominal de rutina se hace el hallazgo incidental de una ectopia renal cruzada, por lo que se deriva al servicio de Urología Pediátrica.
Como antecedentes personales relevantes, fue producto del primer embarazo no mórbido de madre joven. Parto eutócico a las 39 semanas. Tuvo un desarrollo puberal fisiológico con telarquia a los 8 años, adrenarquia a los 9 años, con tasa de crecimiento pondo-estatural dentro de los percentiles para su edad, pero aún no había presentado la menarquia. Destaca dentro de los antecedentes familiares que su madre presentó telarquia y adrenarquia a los 8 años y menarquia los 11 años de edad. Padre joven con un desarrollo puberal aparentemente normal.
El examen físico general de ingreso revela una adolescente sana en buenas condiciones físicas. Las mamas son simétricas, Tanner 5 con complejo areola-pezón desarrollado y de configuración usual. Al examen ginecológico, vello púbico Tanner 5, vulva pequeña (por eje dorsoventral disminuido), sin clitoromegalia. El vestíbulo es corto y con el meato uretral central. El himen se encuentra reemplazado por mucosa gruesa, bien estrogenizada con una pequeña umbilicación ó fosita de 5 mm de profundidad, en lugar de vagina, ubicada dorsalmente al meato uretral central. (Figura 1)
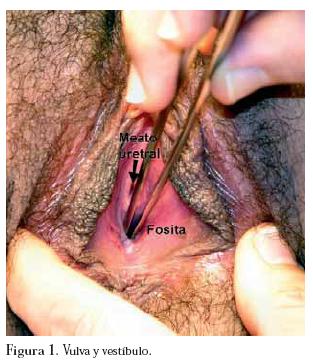
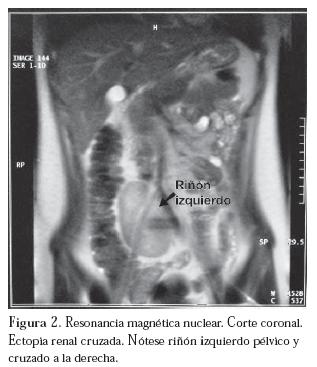
En este punto se plantean los diagnósticos de amenorrea primaria, Síndrome de Mayer-von Rokitansky-Küster-Hauser y ectopia renal cruzada. Se complementa el estudio mediante ecografía abdómino-pélvica que informa un riñón izquierdo no ubicado en la fosa renal sino en el hipogastrio por delante y a la derecha de la columna vertebral. Su forma se adapta al contorno del cuerpo vertebral y mide 8,1 cm de longitud. El riñón derecho es eutópico y de morfología y tamaño normal. A nivel pelviano no es posible identificar útero, ni estructuras müllerianas. Los ovarios son de características puberales usuales.
Se realiza resonancia magnética nuclear (RMN) que confirma los hallazgos ecográficos y el diagnóstico inicial. (Figura 2)
Después de una adecuada evaluación psicológica y multidisciplinaria y por elección de la paciente, se lleva a cirugía, donde se realiza una neovaginoplastia sigmoidea en tres tiempos sucesivos, dentro de la misma intervención, donde además se confirma la presencia de ovarios de características normales y la ausencia de rudimentos Müllerianos.
La primera etapa quirúrgica en posición genupectoral, por abordaje transperineal sagital posterior pre-rectal, mediante una modificación de la técnica de Peña,4,5 con el cual se labra un trayecto desde el periné hasta el fondo de saco de Douglas, dejando una guía en dicho lugar. (Figura 3)
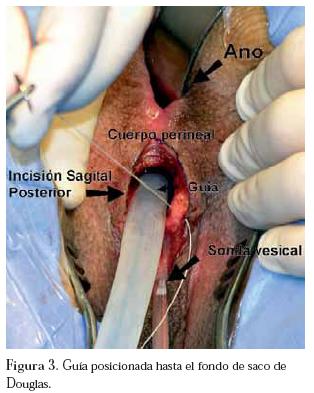
La segunda etapa en decúbito supino, mediante laparotomía mediana infraumbilical, aislando, preparando y rotando 180°, 10 cm de sigmoide con preservación del mesenterio, formando una bolsa ciega, creando así una neovagina sigmoidea, la cual es fijada a la guía transperineal previamente rescatada a través del peritoneo del fondo de saco de Douglas (figura 4) y anastomosis termino-terminal de colon.
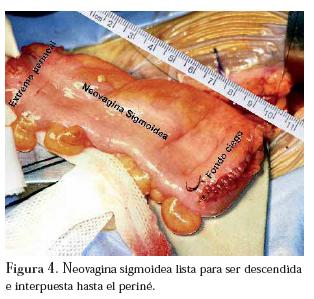
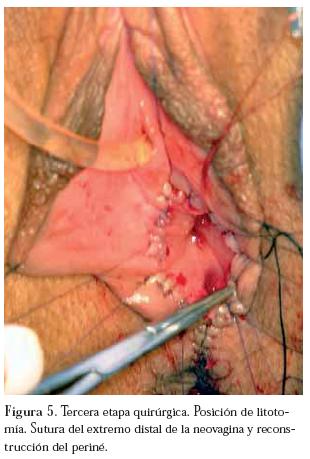
La tercera etapa, en posición de litotomía en la cual se rescata la neovagina por vía perineal y se sutura a la mucosa vaginal circundante y se reconstruye el periné. (Figura 5)
La paciente evoluciona satisfactoriamente en el postoperatorio inmediato dándose alta al tercer día postoperatorio. En controles subsiguientes se verifica patencia y permeabilidad vaginal, con resultados cosmético y funcional excelentes.
DISCUSIÓN
El caso presentado es un buen ejemplo de lo típicamente tardío del diagnóstico de MRKH. Aún cuando no fue nuestro caso, el diagnóstico generalmente se hace en el proceso de un estudio por amenorrea primaria. La edad promedio al diagnóstico es de 15 a 18 años.6,7 Debe, por este motivo, recalcarse la importancia de un examen ginecológico cuidadoso en todas las niñas desde el momento del nacimiento y en casos seleccionados el examen rectal puede ser una herramienta invaluable para descartar la ausencia del útero.5 Por la variabilidad en la longitud del canal vaginal, es importante tener en cuenta que un examen físico en apariencia normal, no descarta por completo el diagnóstico de MRKH.1
El Colegio Americano de Obstetras y Ginecólogos (ACOG)8 recomienda que la primera valoración ginecológica de una niña se haga entre los 13 y 15 años de edad, siendo ésta una inmejorable y temprana oportunidad, además, para instruir a la adolescente y a sus padres en salud reproductiva y hacer intervención a nivel primario, incluyendo problemas del desarrollo sexual y reproductivo, más que enfocado a patología en particular.9
Esta paciente debió haber llamado la atención tiempo antes del momento de la consulta inicial, ya que en primera instancia, su madre había tenido un desarrollo puberal muy similar al suyo, pero con menarquia a los 11 años; y su padre había tenido también un desarrollo puberal aparentemente normal. Adicionalmente, la paciente tuvo un inicio de pubertad a los 8 años y más de 6 años después, a pesar de haber seguido un desarrollo fisiológicamente ordenado, no había presentado menarquia, hecho que es definitivamente anormal, si tenemos en cuenta que el orden le los eventos fisiológicos del desarrollo puberal son, primero aceleración del crecimiento pondo-estatural, seguido por el desarrollo mamario, la adrenarquia y por último la menarquia; esta secuencia de eventos en promedio tarda 4,5 años (1,5 – 6 años). El intervalo promedio entre la telarquia y la menarquia son 2 años y la edad promedio de la menarquia es de 12,8 años. A pesar de que la edad de inicio de la pubertad se está adelantando, no parece ser así para la edad de la menarquia.10 Gaete y colaboradores reportaron un inicio de la pubertad aún más temprano en la población chilena estudiada, con un 14% de niñas, por lo demás sanas, con inicio de la pubertad entre los 7 y 8 años de edad.11
Lo otro que llama la atención, es que el estudio ultrasonográfico inicial no incluye una valoración del contenido pélvico. Es importante ante el hallazgo de malformaciones renales u óseas, buscar malformaciones urogenitales adicionales y viceversa. En MRKH la incidencia de malformaciones mayores de las vías urinarias es ~ 15%. Si incluimos las malformaciones menores, como la ectopia renal, la incidencia aumenta a un ~ 40%. Presentan además, anormalidades esqueléticas en ~ 5 a 10%; generalmente se trata de alteraciones de la columna vertebral: vértebras cuneiformes, fusiones, cuerpos vertebrales rudimentarios y vértebras supernumerarias, aun cuando las extremidades y costillas también pueden estar comprometidas. Raras veces se asocia a anormalidades de los huesos del oído medio y a malformaciones cardíacas.12
La mayoría de las veces es posible llegar al diagnóstico con base en una alta sospecha clínica y un prolijo examen físico y ginecológico, sin embargo, los estudios imagenológicos y la laparoscopia son un complemento valioso para el diagnóstico de anormalidades asociadas y de la morfología precisa de las estructuras pélvicas. La RMN parece ser el mejor método imagenológico para detectar estructuras de la línea media o estructuras dilatadas, pero puede no identificar todos los remanentes müllerianos. En pacientes que se presentan, especialmente con dolor pélvico cíclico la laparoscopia puede ser útil para la evaluación de remanentes uterinos y la etiología del dolor pélvico.7,12
En imágenes de RMN, la agenesia uterina y la hipoplasia pueden ser mejor caracterizadas en los cortes sagitales, mientras que la agenesia vaginal se aprecia mejor en los cortes transversales. La agenesia se demuestra como la no identificación del útero, mientras que la hipoplasia se observa como miometrio con señal de baja intensidad en imágenes en T2, con anatomía zonal pobremente delimitada; la cavidad endometrial y el miometrio se encuentran de tamaño disminuido.13
La agenesia Mülleriana pudiera confundirse con el Síndrome de Insensibilidad Androgénica (SIA), en el cual podría haber también un conducto vaginal corto y ausencia de útero. El SIA es más probable en pacientes con vello púbico y axilar ralo. Se llega al diagnóstico definitivo mediante la determinación del cariotipo, ya que las pacientes con SIA poseen un cariotipo 46XY.12
El tratamiento del MRKH debe ser abordado por un equipo multidisciplinario con experiencia en este tipo de patología. Debe ser precedido por una adecuada evaluación y consejería tanto a la paciente como a sus padres o cuidadores, prestando especial atención a los aspectos psico-sociales, así como a la corrección de la anormalidad anatómica. La cooperación de la paciente, así como su estado mental, son vitales para el éxito de la creación de una vagina funcional.14,15 El momento preciso de la corrección anatómica, bien sea quirúrgico o no quirúrgico, es puramente electivo, por lo que no debe crearse una vagina durante la infancia. Es un reto tanto para la paciente, sus padres y para el clínico, después de la creación quirúrgica de una vagina, el pedirle a la niña que utilice un dilatador vaginal. Estudios de seguimiento a largo plazo han demostrado que las neovaginas creadas durante la infancia tienen una alta tasa de falla y de necesidad de reoperación.16 Si se les da la opción de elegir, algunas pacientes, incluso, optan por nunca someterse a la creación de una neovagina.
La creación de una neovagina se puede lograr tanto por medios quirúrgicos como no-quirúrgicos. El objetivo de cualquier método es el de crear un canal vaginal de un diámetro, longitud y ángulo apropiados para permitir el coito. Adicionalmente, debería tener una secreción normal, suficiente para permitir la lubricación y requerir de un mínimo de cuidados. Desde luego que ninguno de los muchos métodos descritos posee todas estas características juntas, cada uno tiene ventajas y desventajas.6
En el presente caso se optó por la interposición de sigmoide, fundamentado en la elección de la paciente y en las posibles ventajas que en su caso confería, dado que no era una candidata óptima para la creación no quirúrgica de una vagina, ya que el canal vaginal estaba limitado a una diminuta fosita y el vestíbulo era muy corto. Estamos conscientes de que la técnica empleada es compleja y requiere de un entrenamiento adecuado, que incluye un manejo adecuado del colon sigmoide, una gran meticulosidad para lograr preservar la vascularización del asa una vez rotada y descendida y finalmente es necesario conocer a cabalidad la técnica sagital posterior prerectal, ya que existe el riesgo potencial de lesionar uretra, vejiga y recto. A pesar de ser un procedimiento invasivo, el colon sigmoide era una opción óptima, debido a su proximidad al periné, la relativamente fácil movilización de su pedículo vascular y su diámetro apropiado, favoreciendo la utilización de un segmento de tan solo 10 cm de este para la creación de la neovagina. Adicionalmente, tiene las ventajas de no requerir hospitalización prolongada, ni dilataciones posoperatorias, como sucede con la técnica de Abbe-McIndoe. La secreción mucoide tiene sus problemas si se opta por una logitud mayor de intestino, como lo es una mucorrea exagerada, en cambio facilita el coito por la lubricación natural y el resultado cosmético es excelente.
Debido a que actualmente se utilizan segmentos más cortos de colon (8 a 12 cm), la secreción excesiva de moco no es inconveniente. Además, las pacientes y su familia son instruidas para realizar irrigaciones de la neovagina después de la cirugía con suero fisiológico y posteriormente solo de acuerdo a la necesidad. Se ha observado que con el tiempo, la cantidad de secreción disminuye y deja de ser un problema. Por otro lado, ya que los segmentos de intestino utilizados tienen pedículo vascular, además de ser anclados durante el procedimiento, el prolapso es un problema infrecuente. Se favorece también el procedimiento a edades más tempranas.17
Dada la baja prevalencia del MRKH, la cantidad limitada de modelos experimentales y la gran cantidad de aproximaciones terapéuticas y tratamientos quirúrgicos y no quirúrgicos propuestos, es intuitivo concluir que aún no se ha escrito la última palabra acerca del manejo de esta entidad, y son definitivamente necesarios mayores y mejores estudios de seguimiento a largo plazo, que esperamos ver en años venideros, con un poder estadístico suficiente para permitirnos formular recomendaciones veraces basadas en evidencia. Entretanto, sería sensato decir que el MRKH lo debemos tener siempre presente en el estudio de amenorrea primaria, ya que éste es su segunda causa, después de la disgenesia gonadal.6
REFERENCIAS
1. Hickey M, Balen A. Menstrual disorders in adolescence: investigation and management. Hum Reprod Update 2003;9:493-504. [ Links ]
2. Esfandiari N, Claessens EA, O'Brien A, Gotlieb L, Casper RF. Gestational carrier is an optimal method for pregnancy in patients with vaginal agenesis (Rokitansky syndrome). Int J Fertil Womens Med 2004;49:79-82. [ Links ]
3. Ben-Rafael Z, Bar-Hava I, Levy T, Orvieto R. Simplifying ovulation induction for surrogacy in women with Mayer-Rokitansky-Küster-Hauser syndrome. Hum Reprod 1998;13:1470-1. [ Links ]
4. Pena A, Devries PA. Posterior sagittal anorectoplasty: important technical considerations and new applications. J Pediatr Surg 1982;17:796-811. [ Links ]
5. Pena A, Devries PA. Posterior sagittal approach for the correction of anorectal malformations. Adv Surg 1986;19:69-100. [ Links ]
6. Laufer MR. Congenital absence of the vagina: in search of the perfect solution. When, and by what technique, should a vagina be created? Curr Opin Obstet Gynecol 2002;14:441-4. [ Links ]
7. Heller DS. Lower genital tract disease in children and adolescents. J Pediatr Adolesc Gynecol 2005;18:75-83. [ Links ]
8. American College of Obstetricians and Gynecologists. Guidelines for women's health care. 2nd ed. Washington, DC : ACOG; 2002. [ Links ]
9. Breech L, Holland-Hall C, Hewitt G. The well girl exam. J Pediatr Adolesc Gynecol 2005;18:289-91. [ Links ]
10. Lalwani S, Reindollar RH, Davis AJ. Normal onset of puberty have definitions of onset changed? Obstet Gynecol Clin North Am 2003;30:279-86. [ Links ]
11. Gaete X, Unanue N, Ávila A. Cambios en la edad de inicio de la pubertad en niñas de la comuna de Santiago: implicancias para el diagnóstico de la pubertad precoz. Rev Chil Pediatr 2002;73:363-8. [ Links ]
12. Gell JS. Mullerian anomalies. Semin Reprod Med 2003;21:375-88. [ Links ]
13. Troiano RN, McCarthy SM. Mullerian duct anomalies: imaging and clinical issues. Radiology 2004;233:19-34. [ Links ]
14. Poland ML, Evans TN. Psychologic aspects of vaginal agenesis. J Reprod Med 1985;30:340-4. [ Links ]
15. Coney PJ. Effects of vaginal agenesis on the adolescent: prognosis for normal sexual and psychological adjustment. Adolesc Pediatr Gynecol 1992;5:8-12. [ Links ]
16. Goerzen JL, Gidwani GP, Bailez MM, Merritt DF, Caughey S, Yang M. Outcome of surgical reconstructive procedures for the treatment of vaginal anomalies. Adolesc Pediatr Gynecol 1994;7:76-80. [ Links ]
17. O'Connor JL, DeMarco RT, Pope JC 4th, Adams MC, Brock JW 3rd. Bowel vaginoplasty in children: a retrospective review. J Pediatr Surg 2004;39:1205-8. [ Links ]
Conflicto de intereses: ninguno declarado.

