










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Obstetricia y Ginecología
versão impressa ISSN 0034-7434versão On-line ISSN 2463-0225
Rev Colomb Obstet Ginecol v.58 n.4 Bogotá out./dez. 2007
* Patóloga. PhD en Ciencias. Departamento de Patología, Facultad de Medicina, Universidad de Antioquia. Medellín, Colombia. Correo electrónico: condoreduardo@yahoo.com
** Residente, Departamento de Patología, Facultad de Medicina, Universidad de Antioquia. Medellín, Colombia.
RESUMEN
El neuroblastoma congénito es raro, pero es el tumor maligno más común en fetos y neonatos. Produce metástasis entre el 25 y el 50% de los casos. Aunque el pronóstico del neuroblastoma fetal suele ser favorable, incluso en el estadio IV, puede ocurrir muerte fetal o neonatal por hidropesía no inmune. Reportamos un caso de un feto con un neuroblastoma congénito con múltiples metástasis que murió en el útero.
Palabras clave: neuroblastoma, tumor congénito, mortinato.
SUMMARY
Congenital neuroblastoma is rare, but is the most common malignant neoplasm of the foetus and the newborn. Although prognosis remains good in the newborn, in some cases it could lead to non-immune dropsy and intrauterine death. We report a case of congenital neuroblastoma accompanied by multiple metastases found at autopsy performed several days after death. Post-mortem examination revealed a cystic and haemorrhagic mass in the right adrenal and multiple tumour nodules in the lung. Hepatomegaly and placentomegaly were found.
Key words: neuroblastoma, stillbirth, congenital tumour.
INTRODUCCIÓN
El neuroblastoma es raro, pero es el tumor maligno más común en fetos y neonatos y existen algunos reportes de casos en América Latina.1-3 En el momento del diagnóstico se encuentran metástasis entre el 25 y el 50% de los casos.4Aunque el pronóstico del neuroblastoma fetal suele ser favorable, puede ocurrir muerte fetal o neonatal.5 La ecografía obstétrica ha permitido diagnosticarlo in útero desde hace varios años, por lo cual es necesario tenerlo en cuenta en los casos de masa u organomegalia.6 Reportamos un caso de un feto con un neuroblastoma metastásico estadio IV con compromiso placentario e hidropesía que murió en el útero.
DESCRIPCIÓN DEL CASO
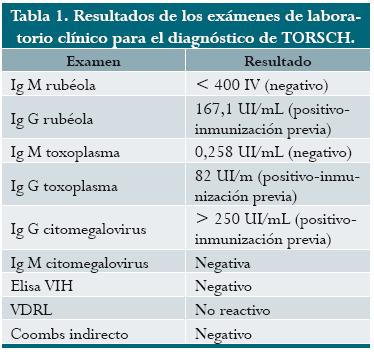
Producto del primer embarazo de una joven sin antecedentes médicos importantes a quien se le practicó control prenatal rutinario en Puerto Berrío, Antioquia. A las 32 semanas de gestación calculadas por ecografía del tercer trimestre, consultó por actividad uterina y fue remitida con diagnóstico de amenaza de parto prematuro e infección. Las pruebas para descartar infección del grupo TORSCH fueron negativas (tabla 1). Se practicó ecografía y se encontró en el feto una masa abdominal. Horas más tarde se halló feto muerto, se inició inducción y por vía vaginal se obtuvo un mortinato, cinco días después.
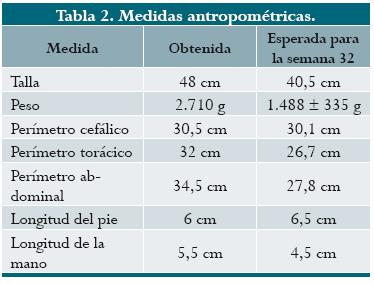
Mortinato de sexo masculino, con maceración generalizada, palidez e hidropesía. Las medidas antropométricas correspondieron a 36 semanas de gestación. Los valores del producto junto con los valores normales7 se describen en la (tabla 2). Se encontraron hidrotórax y ascitis. La placenta, pálida y edematosa, pesó 53% más de lo esperado para la edad gestacional. El examen interno reveló hepatomegalia, múltiples nódulos pulmonares y una masa necrótica y hemorrágica en la glándula suprarrenal derecha. La masa midió 5 x 4 x 3 cm, estaba adherida al riñón y el agregado pesó 52 g.
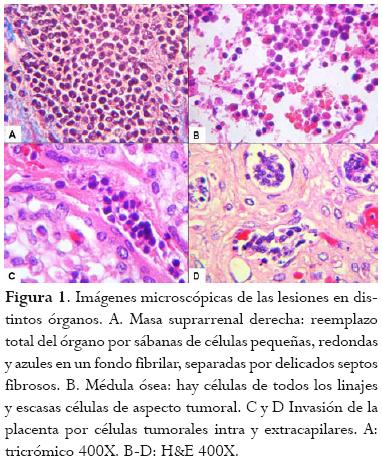
Todos los órganos con alteraciones macroscópicas (ambas adrenales, hígado, pulmones) presentaban infiltración extensa por células pequeñas, redondas y azules en medio de un estroma fibrilar. También se observaron algunas en la médula ósea y en los capilares placentarios (figura 1).
Debido posiblemente a la baja antigenicidad del tejido, el estudio inmunohistoquímico tanto en la masa como en las células que infiltraban los capilares placentarios fue negativo para el antígeno epitelial de membrana, enolasa neuronal específica, cromogranina, antígeno común leucocitario, vimentina y S100. Sin embargo, los hallazgos nos permiten diagnosticar con un grado de certeza razonable un neuroblastoma congénito de estroma pobre, mal diferenciado, en un estadio clínico IV.
Vale la pena mencionar la presencia de células ganglionares en el tejido rectal, lo cual nos permite pensar que no hay asociación con la enfermedad de Hirschprung. No se realizó estudio cromosómico ni búsqueda de la amplificación de N-myc, dado que los costos son elevados en nuestro medio.
La causa de muerte fue posiblemente una descompensación hemodinámica. Hubo falla del ventrículo derecho del corazón, como consecuencia de la obstrucción del drenaje venoso derecho, la cual interfirió con la circulación fetoplacentaria.
Este fenómeno se originó en la infiltración masiva del hígado por las células cancerosas.
DISCUSIÓN
Aunque las neoplasias congénitas son raras, deben tenerse en cuenta como parte del diagnóstico diferencial en casos de masa, organomegalia e hidropesía. Además, como parte del diagnóstico diferencial de la placentomegalia deben considerarse los tumores congénitos además de la infección, la diabetes materna y la eritroblastosis fetal.1,8
Los tumores congénitos malignos más comunes son los tumores de células germinales, la leucemia congénita o síndrome mieloproliferativo transitorio, el neuroblastoma y el hepatoblastoma.9,10 El neuroblastoma es la tercera neoplasia pediátrica después de la leucemia y los tumores del sistema nervioso central.11Todos ellos pertenecen al grupo de los blastomas y parecen ser defectos del desarrollo de los órganos.8,9
Los neuroblastomas se originan en el sistema nervioso simpático y se desarrollan en cualquier lugar donde este exista. Los sitios más comunes son la suprarrenal (35%), el retroperitoneo (30-35%), y el mediastino posterior (20%). La mayoría son esporádicos, pero algunos casos pueden ser familiares.12 Se ha reportado asociación del neuroblastoma con la enfermedad de Hirschsprung, la enfermedad de von Recklinghausen, el síndrome de Beckwith-Wiedemann y con el síndrome de Ondina.12,13 Su comportamiento es agresivo, y forma parte de casi el 15% de las muertes por cáncer en la infancia; sin embargo, en ocasiones se ha detectado regresión espontánea o su maduración a lesiones benignas.11
Los sitios más comunes de metástasis por neuroblastoma son el hueso, la médula ósea, el hígado, la placenta y la piel.7,11,14 En este caso, el patrón de metástasis es bastante típico. En el caso de neuroblastoma, las metástasis pulmonares y cerebrales son raras y en el caso de otros tumores el compromiso placentario es inusual.13
El neuroblastoma congénito es el tumor más común durante el primer mes de vida, siendo un 30-50% de todas las neoplasias a esta edad. Este está dividido en neuroblastoma neonatal y fetal, basado en la edad de presentación. Ambos son de buen pronóstico, pero difieren en el patrón de metástasis y el órgano de origen. El neonatal tiene en un 45% origen en la suprarrenal y un 60% tiene metástasis en el momento del diagnóstico; tiene generalmente un pronóstico favorable. El neuroblastoma fetal es diagnosticado por ultrasonido a una edad gestacional media de 36 semanas. En el 90% es de origen en la suprarrenal, se asocia con metástasis hepática y la llamada metástasis placentaria se limita a los vasos sin comprometer el parénquima, dando origen a hidropesía fetal. En algunas ocasiones se puede producir preeclampsia debido a la secreción de catecolaminas.11,12
Los neuroblastomas han sido clasificados según dos sistemas histológicos, los cuales se han usado para establecer grupos de riesgo y pronóstico: la clasificación de Shimada y la clasificación del grupo de oncología pediátrica americano (POG)15 (tabla 3).

Se han usado una variedad de marcadores para complementar el diagnóstico de neuroblastoma, los cuales también sirven como marcadores de seguimiento y pronóstico. Los más convencionales son los metabolitos urinarios de las catecolaminas ácido vanilmandélico VMA y HVA y la dopamina. Hay otros marcadores como la ferritina, la deshidrogenasa láctica o LDH, enolasa específica de neurona, GD2 entre otros. De todos ellos, solo las catecolaminas son características y están lo suficientemente estandarizados. La ferritina y la LDH son útiles en el pronóstico, pero son poco sensibles y específicos. La enolasa específica de neurona y el GD2 séricos son específicos pero no tan sensibles y no siempre están disponibles.16 Debido a la investigación continua, han aumentado los marcadores genéticos relacionados con el pronóstico del neuroblastoma. Se han identificado anormalidades en el cariotipo; la más común es la ganancia en el brazo largo del cromosoma 17, la cual está asociada con un pronóstico adverso. La deleción del cromosoma 1 ocurre en 30 a 50% de los tumores primarios y se correlaciona fuertemente con la amplificación del gen N-myc y un pobre pronóstico.16
La categorización clínica está fundamentada en el sistema de estadificación internacional para neuroblastoma (INSS) (tabla 4). Esta clasificación está basada en el comportamiento tumoral luego de la resección y el compromiso de ganglios linfáticos, sin importar la presencia de enfermedad residual microscópica.

Los protocolos de tratamiento se han designado de acuerdo con la estratificación del riesgo de la lesión (bajo, intermedio y alto) según la edad, el estadio y las características biológicas (amplificación de N-myc, ploidía e histopatología).10 Se consideran de bajo riesgo los pacientes en los estadios 1, 2a o 2b sin amplificación de N-myc, los que tienen histología favorable con amplificación de N-myc, los recién nacidos estadio 2 sin importar la amplificación, como también los neonatos estadio 4S sin amplificación de N-myc, debido a sus altas tasas de regresión espontánea y supervivencia.16 En el presente caso, la determinación de la amplificación de N-myc hubiese sido de utilidad; sin embargo, la sobrevida de los niños en estadio 4 es del 20 al 35%.17,18
Los pacientes de bajo riesgo tienen excelente respuesta con el tratamiento quirúrgico. Los de riesgo intermedio tienen un pronóstico excelente, con una sobrevida del 90% luego de un curso de quimioterapia con cisplatino, etopósido, ciclofosfamida y doxorrubicina. Para los de alto riesgo, generalmente el tratamiento consiste en inducción con quimioterapia, terapia mieloablativa a altas dosis, trasplante alogénico o autólogo de médula ósea o de células madre, cirugía y en algunos casos radiación.17
Con la aparición de los estudios ecográficos de alta resolución se diagnostican cada día más casos congénitos. Aunque por medio de una ecografía es difícil distinguir un neuroblastoma de una hemorragia suprarrenal o de alteraciones renales, debe sospecharse en el caso de un feto con una masa suprarrenal quística o sólida, de pared gruesa, acompañada o no de hepatomegalia o lesiones hepáticas.11,19Las hemorragias suprarrenales no son comunes en los fetos8 y en caso de sospecha durante el embarazo, o si se encuentra hepatomegalia, placentomegalia o hidropesía fetal, debe considerarse la terminación del embarazo.19
Como se mencionó anteriormente, la mayoría de los niños con neuroblastoma congénito tienen un pronóstico favorable. Algunos de ellos se observan durante semanas o meses antes de instaurar medidas terapéuticas, dado que muchos de ellos desaparecen o disminuyen de tamaño con el tiempo;17 el tratamiento para seguir depende de la clasificación del riesgo.
Es posible que ante el crecimiento de nuestras instituciones y la introducción de nuevos servicios, el diagnóstico de casos como este sea más frecuente, por lo cual, el conocimiento de estas entidades y su pronóstico son necesarios.
REFERENCIAS
1. Smith CR, Chan HS, deSa DJ. Placental involvement in congenital neuroblastoma. J Clin Pathol 1981;34:785-9. [ Links ]
2. Palomino-Dávila VP, Carbajal-Chávez T. Neuroblastoma Congénito. VII Congreso Virtual Hispanoamericano de Anatomía Patológica 2005. Visitado 2007 may 31. Disponible en: http://www.conganat.org/7congreso/trabajo.asp [ Links ]
3. Bastián Manso B, Agüero-Díaz A. Hidrops fetalis y neuroblastoma congénito: presentación de 1 caso. Rev Cuba Pediatr 1987;59:667-70. [ Links ]
4. Castilla EE, Orioli IM. ECLAMC: The Latin-American collaborative study of congenital malformations. Community Genet 2004;7:76-94. [ Links ]
5. Luis AL, Martínez L, Hernández F, Sastre A, García P, Queizan A, et al. Congenital neuroblastomas. Cir Pediatr 2004;17:89-92. [ Links ]
6. Acharya S, Jayabose S, Kogan SJ, Tugal O, Beneck D, Leslie D, et al. Prenatally diagnosed neuroblastoma. Cancer 1997;80:304-10. [ Links ]
7. Kalousek DK, Baldwin VJ, Dimmick JE, Norman MG, Cimolai N, Andrews A, Paradice B. Embryofetal-perinatal autopsy and placental examination. En: Dimmick JE, Kalousek DK, eds. Developmental pathology of the embryo and fetus. Philadelphia East Washington Square: JB Lippincott Company; 1992. p. 761-99. [ Links ]
8. Ruiz Villaespera A, Suarez Mier MP, López Ferrer P, Álvarez Baleriola I, Rodríguez González JI. Nonimmunologichydropsfetalis:anetiopathogenetic approach through the postmortem study of 59 patients. Am J Med Genet 1990;35:274-9. [ Links ]
9. Keeling JW. Hydrops fetalis and other forms of excess fluid collection in the fetus. En: Wigglesworth JS, Singer DB,eds. Textbook of fetal and perinatal pathology. Boston: Blackwell Scientific Publications; 1991. p. 429-54. [ Links ]
10. Debich-Spicer D. Congenital tumors. En: Gilbert-Barness, Debich-Spicer D, eds. Embryo and fetal pathology: color atlas with ultrasound correlation. New York: Cambridge University Press; 2004. p. 546-78. [ Links ]
11. Papaioannou G, McHugh K. Neuroblastoma in childhood: review and radiological findings. Cancer Imaging 2005;5:116-27. [ Links ]
12. Lonergan GJ, Schwab CM, Suárez ES, Carlson CL. Neuroblastoma, ganglioneuroblastoma and ganglioneuroma: radiologic-pathologic correlation. Radiographics 2002;22:911-34. [ Links ]
13. Trochet D, O’Brien LM, Gozal D, Trang H, Nordenskjöld A, Laudier B,etal. PHOX2Bgenotype allowsforprediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet 2005;76:421-6. [ Links ]
14. Sul HJ, Kang D. Congenital neuroblastoma with multiple metastases: a case report. J Korean Med Sci 2003;18:618-20. [ Links ]
15. Joshi VV, Rao PV, Cantor AB, Altshuler G, Shuster JJ, Castelberry RP. Modified histologic grading of neuroblastomas by replacement of mitotic rate with mitosis karyorrhexis index. A clinicopathologic study of 223 cases from the Pediatric Oncology Group. Cancer 1996;77:1582-8. [ Links ]
16. Broudeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993;11:1466-77. [ Links ]
17. Henry MC, Tashjian DB, Breuer CK. Neuroblastoma update. Curr Opin Oncol 2005;17:19-23. [ Links ]
18. Le Bouar G, Lassel L, Loeuillet-Olivo L, d’Herve D, Poulain P. Antenatal metastatic neuroblastoma: prognostic criteria. A case report. J Gynecol Obstet Biol Reprod 2003;32:740:4. [ Links ]
19. Deeg KH, Dachert C, Glöckel U, Langer T. The different features of congenital neuroblastomas: report about the sonographic diagnosis of three cases Ultraschall Med 2007;28:313-20. [ Links ]
Consideraciones éticas: los padres aceptaron, a través de a firma de un consentimiento informado, la realización de a autopsia fetal según los protocolos del HSVP para dicho procedimiento. Se garantiza la confidencialidad de los sujetos.
Conflicto de intereses: ninguno declarado.

