











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
Los tumores de Ewing comprenden numerosas variedades histológicas clasificadas en subtipos, entre las que se encuentran: el sarcoma de Ewing óseo y de tejidos blandos (90 % de los casos), el tumor neuroectodérmico primitivo periférico, el tumor de Askin (tumor neuroectodérmico primitivo de la pared torácica), y otras variantes menos frecuentes. Se trata de un tumor maligno constituido por una proliferación uniforme de células pequeñas y redondeadas, que no producen matriz extracelular y presentan una diferenciación neuroectodérmica parcial. Se caracterizan por presentar fusiones génicas que incluyen al gen EWS (Ewing sarcoma) del cromosoma 22 y a algún gen de la familia ETS de factores de transcripción. Solo en 1 de cada 10 pacientes se localiza en tejidos extraóseos 1,2.
Los sarcomas de Ewing cérvico-vaginales se observan de forma excepcional, presentando una variedad histológica amplia. Constituyen menos del 1 % de las lesiones malignas cervicales, y se dividen en subtipos tales como el rabdomiosarcoma embrionario, leiomiosarcoma, sarcoma endocervical indiferenciado, sarcoma alveolar de partes blandas y liposarcoma 3. Su comportamiento es muy agresivo, ya que presenta una alta tasa de recidivas si el diagnóstico es tardío, localizándose principalmente en el tórax, las extremidades y el retroperitoneo, aunque debido al escaso número de artículos en los que se hace referencia a estas, no existen porcentajes consensuados 4,5.
Los tumores de Ewing tienen una incidencia aproximada de 3 casos/millón de habitantes/año, y representan cerca del 3-6 % de los tumores sólidos y el 1,4- 1,8 % de los procesos malignos, presentándose generalmente en la segunda década de vida 6. La prevalencia de las formas extraóseas de variedad cérvico-vaginal es de 0,2 pacientes por millón de habitantes 4, es uno de los subtipos considerados extremadamente raros 6-8. El diagnóstico se sospecha fundamentalmente por la presentación clínicay se confirma con el estudio histológico, inmunohistoquímico y genético. No existe actualmente prueba de imagen capaz de discernir entre las numerosas variedades 3. Existen tres tratamientos esenciales: la cirugía, la radioterapia y la quimioterapia 4. Por otro lado, el pronóstico depende tanto del subtipo como de la capacidad metastásica del tumor.
A continuación se presenta el caso de una paciente diagnosticada con sarcoma de Ewing extraóseo ubicado en el tracto genital inferior. El objetivo de presentar este caso es realizar una revisión de la literatura respecto al diagnóstico clínico, inmunohistoquímico, molecular y genético, y sobre la seguridad y eficacia de las diferentes estrategias terapéuticas para su manejo en la actualidad.
REPORTE DE CASO
Paciente de 53 años, nulípara, con dos abortos previos y antecedente personal de cáncer de mama ductal infiltrante a los 50 años de edad, intervenida mediante cirugía conservadora de mama izquierda y linfadenectomía axilar ipsilateral, en tratamiento con tamoxifeno (20 mg/día), por 3 años, al momento en que es remitida a consulta de ginecología general tras presentar sangrado genital anormal en varias ocasiones.
La paciente es atendida en el Hospital Universitario de Jerez de la Frontera, institución hospitalaria general pública de referencia en la provincia de Cádiz. En entrevista describe el sangrado como ocasional, escaso y sin motivo relacionado aparente. Durante la exploración ginecológica se visualiza un sangrado escaso de cavidad, observándose a nivel cervical y de fondo de saco vaginal derecho una lesión excrecente sospechosa de malignidad, de la cual se toma biopsia. En el tacto bimanual se palpa una masa pélvica mayor de 20 cm que se extiende por encima de ombligo, con útero de movilidad reducida. Se realiza una ecografía vía vaginal en la que se visualiza una tumoración en cérvix de 5,5 cm, abigarrada y que capta Doppler color. Por otro lado, a través de esta prueba de imagen se confirma la presencia de una tumoración pélvica uterina de 20 x 10 cm (descrita durante el tacto bimanual), que ocupa todo el abdomen inferior de la paciente, que puede corresponder a un proceso infiltrativo de la propia masa cervical. Se intenta realizar una biopsia endometrial, siendo técnicamente imposible por lo que se indica como mejor opción diagnóstica en ese momento una histeroscopia ambulatoria. Se realiza el procedimiento sin complicaciones, apreciándose una cavidad tortuosa a expensas de pequeños nódulos con hipervascularización anómala, sobre todo en fondo uterino. Ante dichos hallazgos se realiza microlegrado endocavitario, sin obtener muestra suficiente para confirmar el diagnóstico anatomopatológico. Para completar el estudio se solicita una resonancia nuclear magnética en la que, como hallazgo principal, se observa un útero tumoral aumentado de tamaño, con una extensión longitudinal de 20 cm y un diámetro anteroposterior aproximado de 10 cm, con lesión de aspecto infiltrativo que ocupa totalmente la cavidad endometrial, e infiltración de la totalidad del miometrio. También se visualiza extensión a pelvis, parametrios, fondos de sacos vaginales y tercio superior de vagina. El estudio radiológico revela grandes adenopatías sospechosas de malignidad retroperitoneales y pélvicas, de 22 mm en el hilio renal izquierdo, intercavo-aórtica de 19 mm, iliacas comunes bilaterales y bifurcación bilateral de iliacos externos e internos, la mayor de 30 mm en bifurcación de iliacos externos e internos derechos. Se observa presencia de líquido libre loculado intraperitoneal en fondo de saco de Douglas en pequeña cuantía, y ovarios desplazados en dirección craneal, siendo estos de pequeño tamaño y sin alteraciones macroscópicas. No se identifica afectación de vejiga ni de rectosigma. Considerando todos estos estudios, el diagnóstico de sospecha nos orienta hacia una neoplasia cervical, en espera de confirmación. El resultado anatomopatológico de la lesión biopsiada, que engloba cérvix y fondo de saco anterolateral de vagina, muestra hallazgos compatibles con sarcoma de células redondas tipo Ewing, o tumor neuroectodérmico primitivo. El estudio inmunohistoquímico resulta positivo para vimentina y CD99, y negativo para marcadores epiteliales y linfoides, actina, desmina, S100, marcadores neuroendocrinos y CD10. Por ello, se concluyó como juicio clínico principal: infiltración masiva de estructuras pélvicas y ganglionares, por sarcoma de células redondas compatible con sarcoma de Ewing o tumor neuroectodérmico primitivo cervical. Tras comentar el caso en el Comité Oncológico de tumores, se decide iniciar tratamiento médico quimio-radioterápico por parte del servicio de Oncología, ante la imposibilidad de resección quirúrgica óptima. El tratamiento elegido fueron ciclos de ciclofosfamida, doxirrubicina y vincristina, alternados con ifosfamida, etopósido y radioterapia. Actualmente, la paciente continúa con el tratamiento de quimioterapia, en seguimiento por el servicio de Oncología, y en espera de valorar la respuesta al mismo.
MATERIALES Y MÉTODOS
Se realizó una búsqueda bibliográfica en las bases de datos Medline vía PubMed, utilizando los términos: "sarcoma; ewing", "neuroectodermal tumors", "primitive", "peripheral" y "uterine cervical neoplasms", en artículos de revisión bibliográfica, editoriales, reportes y series de casos clínicos que describieran la localización genital o pélvica, en inglés y español, publicados en los últimos 10 años. Por otro lado, se excluyeron aquellas publicaciones en las que no se hicieran referencias importantes a las localizaciones tumorales extraóseas, además de estudios en animales.
Aspectos éticos. Se solicitó autorización tanto a la paciente como al Comité de ética en Investigación del HU de Jerez de la Frontera (Cádiz), para la publicación de las imágenes y la información referente al proceso clínico. Se garantizó la confidencialidad tanto de la información como de la identificación de la paciente, para proteger sus derechos de privacidad.
RESULTADOS
Se identificaron 62 artículos, incluyendo finalmente 12 publicaciones que cumplieron con los criterios de búsqueda 1-12. Se excluyeron 50 artículos por no tener referencias lo suficientemente extensas de las formas extraóseas / neuroectodérmicas primitivas. Los artículos seleccionados corresponden a revisiones de la literatura 1, reporte de casos o series de casos clínicos 2,4-6,8-12, editoriales 3 y revisiones de la literatura a partir de casos clínicos (7). Todos los artículos son publicaciones en inglés 1-3,5,7-12, excepto dos, que están escritos en español 4,6.
Diagnóstico. La sospecha clínica se produce con la consulta de la paciente por un tipo de sangrado genital anormal o por la presencia de una tumoración cérvico-vaginal en la exploración. Otras manifestaciones están relacionadas con el crecimiento tumoral y la consecuente compresión de estructuras adyacentes, generando cuadros de dolor intenso. En nuestra revisión observamos 5 casos de sangrado genital o coitorragia 2,7,9,11,12, 4 casos de dolor abdominal 2,4,8,9, aparición de masas de localización genital o pélvica en 7 casos 4-6,8,11,12 y, por último, síntomas derivados de lesiones metastásicas de localización cerebral o pulmonar en 3 pacientes 5,8,9. La edad más frecuente de aparición de los tumores neuroectodérmicos primarios cervicales se encuentra entre los 20 y los 50 años, y varía en otras localizaciones donde se suelen diagnosticar en edades más precoces 6,8. En nuestro caso, la paciente tiene 53 años cuando se diagnostica de sarcoma de Ewing extraóseo.
Diagnóstico histopatológico e inmunohistoquímico / molecular. El análisis inmunohistoquímico es el método principal de diagnóstico, siendo la detección del CD99 y la tinción de vimentina las formas más características y de mayor sensibilidad en este tipo de tumores. El sarcoma de Ewing se caracteriza histológicamente por la aparición de células pequeñas, azuladas, con núcleo redondeado, con positividad para la glicoproteína de membrana p30/32MIC2 (CD99), y con anticuerpos monoclonales que incluyen O13, 12E7 y HBA71 6. Las formas extraóseas de este tipo de tumores son muy infrecuentes, caracterizándose, desde el punto de vista histopatológico, por el desarrollo celular uniforme, con nucleolos prominentes, depósitos de glucógeno y presencia de rosetas 4. La base diagnóstica de los tumores de Ewing extraóseos se basa en la morfología histopatológica, el examen inmunohistoquímico y el estudio genético de la muestra biológica 8 (figura 1). En ocasiones, los marcadores epiteliales y la desmina han podido mostrar positividad focal, mientras que la expresión de marcadores neuronales tales como NSE, S-100, neurofilamentos, proteína ácida fibrilar glial y CGA obtienen resultados más inciertos 1,2,9,10. En nuestra paciente, los marcadores epiteliales y linfoides, actina, desmina, S100, marcadores neuroendocrinos y CD10 resultaron negativos; sin embargo, la tinción de vimentina y glicoproteína de membrana CD99, fueron positivas (figuras 2 y 3). En nuestra revisión hubo 8 casos positivos para CD99 2,5-8,10,12 y 7 para vimentina 2,5,6,8,10,11; por otro lado, resultaron positivos algunos marcadores más inespecíficos como NSE 2, CD 56 5,7, CD117 2,6, CD10 10 o desmina 10.
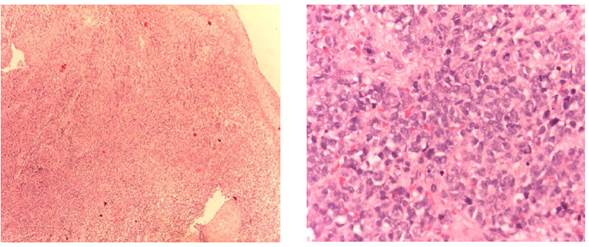
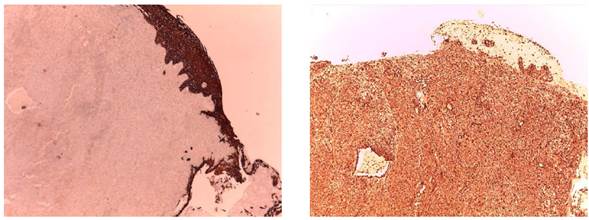
Figura 2 Primer corte con tinción CK negativa. El segundo corte muestra tinción de vimentina positiva
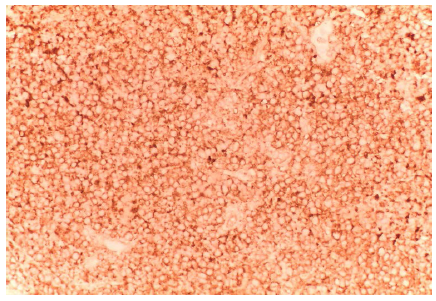
Diagnóstico genético. El sarcoma de Ewing y los tumores neuroectodérmicos primitivos son miembros de una familia de neoplasias asociadas a una translocación cromosómica, específicamente del brazo largo de los cromosomas 11 y 22 expresados como t(q24; q12) o (q22; q12), resultando enla fusión de genes FLI-1 y EWS o EWS/ERG y sobreexpresión de MIC-2/CD99 en la membrana de las células tumorales 1,2. La translocación cromosómica característica (Q24; q12), resultante de la fusión EWS-FLI1, se observa en el 85 % de los casos, mientras que la mayoría de los casos restantes (10-15 %) muestran la fusión de los genes EWS-ERG, que se produce como consecuencia de la translocación cromosómica t(21; 22) (q22; q12) 11. En nuestra revisión observamos dos casos de pacientes con estudio genético positivo para los genes EWSR1 7 y EWS-FLI1 y 2 11.
Diagnóstico mediante técnicas de imagen. El diagnóstico de este tipo de tumores se basa en la adecuada exploración clínica asociada a pruebas de imagen para estudio de extensión, siendo la tomografía axial computarizada (TAC) y, sobre todo, la resonancia nuclear magnética (RNM) las técnicas de elección 4. Nuestra búsqueda bibliográfica y análisis de los artículos seleccionados, mostró que en cinco casos se solicitó RNM 2,5,7,8,11, en seis ecografía abdomino-pélvica 4,8,10-12, en seis casos TAC 4-6,8,10,12 y en dos casos tomografía por emisión de positrones (PET/TAC) 7,10, tanto para el diagnóstico como para el seguimiento posterior al tratamiento.
Estrategias terapéuticas. El tratamiento de los tumores de Ewing presenta tres pilares fundamentales independientemente del tipo histológico o la localización que presente, teniendo en cuenta el diagnóstico precoz de la lesión: la cirugía (como método locorregional, logrando amplios bordes de resección libres de enfermedad), la quimioterapia (ciclos administrados con agentes alquilantes como doxirrubicina, ciclofosfamida o ifosfamida, y otros agentes citostáticos como actinomicina D, vincristina y etopósido), y radioterapia a altas dosis, cuya finalidad es conseguir una evolución favorable y una baja incidencia de recidiva 1,8. Existe la opción de iniciar un tratamiento neoadyuvante quimio-terapéutico previo a la cirugía, con el objetivo de reducir el volumen tumoral, realizado sobre todo en lesiones extensas en las que no se pueda asegurar la extirpación óptima inicial, continuándose posteriormente con nuevos ciclos de quimioterapia y radioterapia tras la extirpación del sarcoma 2,11,12. En nuestra paciente, el tratamiento de elección ha sido la quimioterapia neoadyuvante basada en ciclos con ciclofosfamida, doxirrubicina y vincristina, alternados con ifosfamida, etopósido y radioterapia a altas frecuencias, ya que en el comité de tumores se catalogó este tumor como irresecable quirúrgicamente. En el caso de los tumores sin metástasis, la resección quirúrgica combinada con quimioterapia es el tratamiento de elección 2,8,12. El uso de agentes quimioterapéuticos para el tratamiento de las formas localizadas mejora enormemente la tasa de supervivencia en 5 años de un 5-10 % inicial hasta un 65 %, debido a la eliminación de las posibles micrometástasis existentes. Las pacientes con metástasis a distancia en el momento del diagnóstico, como es el caso que presentamos, desarrollan una menor respuesta al tratamiento y una tasa de recaídas mucho mayor, presentando un peor pronóstico de su enfermedad 6. Existe muy poca literatura sobre el manejo del tumor de Ewing extraóseo, por tratarse de una lesión extremadamente rara, ya que los análisis observacionales a gran escala para protocolizar el manejo de este tipo de pacientes son casi inexistentes, ante la imposibilidad de obtener una muestra elevada para su estudio. Por ello, actualmente no disponemos de datos suficientes para asegurar si en este tipo de tumores localizados es factible iniciar quimioterapia y posteriormente cirugía, o radioterapia, y así conseguir unos resultados terapéuticos óptimos, como procede en los tumores de Ewing localizados en hueso 4. En nuestra revisión, 4 pacientes recibieron el triple tratamiento con cirugía, quimio y radioterapia 6,7,10,12, los demás recibieron diferentes combinaciones de tratamiento: cinco pacientes quimioterapia 2,4,5,8,11, tres radioterapia 2,5,11 y cuatro cirugía 4,8,9.
CONCLUSIONES
El tumor de Ewing extraóseo, o tumor neuroectodérmico periférico primitivo, es una lesión extremadamente rara, más aún en localizaciones de origen ginecológico, como la cervical o vaginal. El diagnóstico se basa, principalmente, en el estudio de la morfología histopatológica, el examen inmunohistoquímico y las pruebas genéticas, siendo el análisis inmunohistoquímico el medio de diagnóstico de mayor efectividad. Desde el punto de vista terapéutico, el diagnóstico precoz del sarcoma es el principal factor de supervivencia, asociado al manejo multidisciplinar, en el que se incluye la combinación de cirugía, quimioterapia y radioterapia.

