










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.29 no.1 Bogotá Jan./Mar. 2000
ARTÍCULO DE REVISIÓN
LA NORADRENALINA SU ROL EN LA DEPRESIÓN
NORADRENALINE ITS ROLE IN DEPRESSION
JORGE TELLEZ VARGAS*
Jefe Departamento de Psiquiatría. Fundación Universitaria de Ciencias de la Salud. Hospital San José, Bogotá. Vicepresidente Federación Latinoamericana de PsiquiatrÃa Biológica. Director Científico de la Asociación Colombiana contra la Depresión y el Pánico.
E-mail: tellezjorge@hotmail.com.
RESUMEN
A partir de las investigaciones realizadas en los años 60 y la respuesta al tratamiento con antidepresivos tricíclicos se ha involucrado a la noradrenalina en la etiología del episodio depresivo mayor. La aparición de los inhibidores selectivos de la recaptación de serotonina, con igual eficacia y menor incidencia de efectos secundarios que los antidepresivos tricíclicos impulsó y desarrolló la etiología serotoninérgica en los trastornos afectivos. Investigaciones recientes y diseño de moléculas antidepresivas, como la reboxetina, consideradas inhibidoras selectivas de la recaptación de noradrenalina (NARI), han puesto nuevamente de relieve los aspectos noradrenérgicos en la etiología y terapéutica de los episodios depresivos.
El presente artículo revisa los aspectos biomoleculares, los modelos de investigación animal y la farmacodinámica de las nuevas moléculas antidepresivas de tipo noradrenérgico.
Palabras Clave: Noradrenalina, depresión, antidepresivos, reboxetina.
ABSTRACT
From investigation in the 60's and therapeutic response to tricyclic antirepressants, naradrenaline has been involved in the etiology of major depressive episodes. Introduction of selective serotonin reuptake inhibitors, equally efficientbut with lesser incidence of secondary effects than tricyclics, favored serotoninergic etiology in the affective disorders. Recent investigations and synthesis of antidepressive molecules, such as reboxetine, selective noradrenaline reuptake inhibitors (NARI), newly emphasizes the role of noradrenaline in etiology and therapy of depressive episodes.
This paper reviews biomolecular aspects, animal investigation models and pharmacodynamics of noradrenergic antidepressive molecules.
Key Words: Noradrenaline, Depression Disorder, Antidepressants, Reboxetine.
INTRODUCCIÓN
Schildkraut, Bunney y Davis postularon en 1965 la teoría que invocaba el déficit de noradrenalina como la causa de la enfermedad depresiva, trabajo que constituyó el primer intento de explicación biológica del síndrome depresivo(1,2). Con base en estos postulados se diseñó el tratamiento farmacológico con inhibidores de la monoamino oxidasa (IMAOS) y los antidepresivos tricíclicos, fármacos que parcialmente respaldaron la teoría noradrenérgica porque actúan en forma simultánea sobre los receptores noradrenérgicos, serotoninérgicos e histaminérgicos.
La noradrenalina es un neurotransmisor que se ha relacionado con la motivación, el estado de alerta y vigilia, el nivel de conciencia, la percepción de los impulsos sensitivos, la regulación del sueño, del apetito y de la conducta sexual y la neu-romodulación de los mecanismos de recompensa, aprendizaje y memoria, funciones que con frecuencia se encuentran alteradas en el paciente deprimido. A excepción de la vigilancia, las demás funciones involucran la acción conjunta de la noradrenalina y otro neurotransmisor (Figura 1).
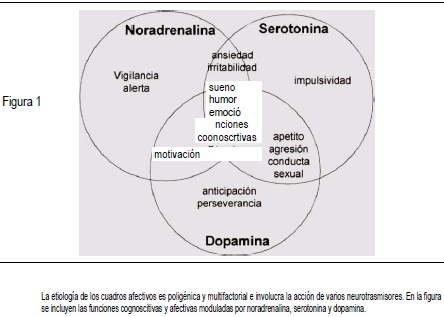
La disminución en la concentración de noradrenalina encontrada en los pacientes con trastornos de la conducta alimentaria parece ser la responsable de la hipotensión arterial, la bradicardia, la hipotermia y la depresión que se observa en este grupo(3).
ASPECTOS NEUROANATÓMICOS
Las neuronas noradrenérgicas están localizadas en el locus ceruleus, situado en el piso del cuarto ventrículo, y en el área tegmental lateral y desde allí proyectan sus conexiones al tálamo, la amígdala, el hipocampo, el hipotálamo y la corteza cerebral (neocortex). El locus ceruleus juega un papel preponderante en las respuestas de adaptación y vigilancia preparando al individuo para la lucha o la huida, coordinando las respuestas al estrés y al miedo (Figura 2).
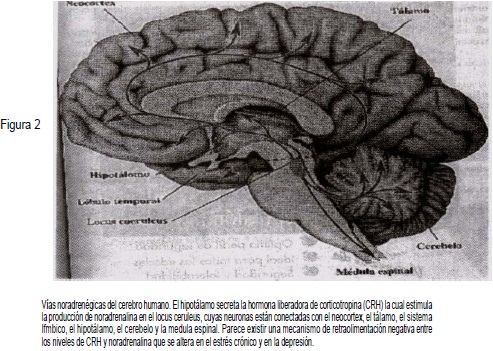
El locus ceruleus actúa como un sistema fásico que mantiene el estado de vigilia y prepara al animal o al individuo para luchar o huir. Este sistema puede desincronizarse y originar alteraciones a nivel simpático central y periférico, disfunción que constituiría las bases neuroana-tómica y neurofísiológica de los trastornos psiquiátricos relacionados con el estrés, como la depresión mayor o el trastorno de estrés post-traumático.
Con la edad disminuyen el número de las células del locus ceruleus, los receptores y la producción de noradrenalina.
El área tegmental integra las funciones autonómicas a través de las neuronas preganglionares sinápticas y los núcleos del tracto solitario y el motor dorsal del vago. Su activación produce bradicardia e hipotensión arterial.
La hormona liberadora de corticotropina (CRH) actúa como un neurotransmisor sobre las neuronas del locus ceruleus, estimulando la producción de noradrenalina, la cual por un mecanismo de retroalimen-tación inhibe la CRH.
METABOLISMO DE LA NORADRENALINA
La noradrenalina es sintetizada a partir del aminoácido fenüalanina, el cual por acción de la fenilalanina hidroxüasa y el cofactor pteridina es convertido en tirosina, que por acción de la tirosina hidroxüasa y la presencia del ion ferroso, de oxígeno y del cofactor tetrahidropteridina, es trasformada en dopa.
La dopa sufre pérdida de un átomo de carbono, por acción de la dopa decarboxilasa y es trasformada en dopamina, que por acción de la dopamina beta hidroxüasa en presencia de cobre, oxígeno y fosfato ascórbico es convertida en la noradrenalina (Figura 3).
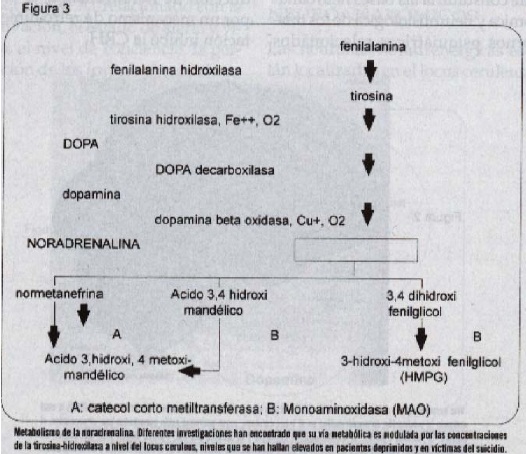
El metabolismo de la noradrenalina sigue tres vías. Una parte es trasformada en normetanefrina, que por acción de catecol-orto-metil-trasferasa (COMT) es convertida en el ácido 3-hidroxi-4-metoxi-mandélico. Otra porción es trasformada en ácido 3-4 dihidroximandélico, que por acción de la monoamino oxidasa (MAO) es convertido en ácido 3-hidroxi-4-metoxi mandélico, y otra es trasformada en 3,4 dihidroxi-fenil-glicol que por acción de la MAO es metabolizado a 3-hidroxi-4-metoxi-fenil glicol (HMPG) el metabolito más importante de la noradre-nalina(4).
A partir del estudio pionero de Maas(5) diferentes investigadores han utilizado los niveles séricos o urinarios del HMPG corno un marcador biológico de la actividad no-radrenérgica y un predictor de la respuesta antidepresiva.
Teniendo en cuenta los hallazgos de Maas se supuso que los pacientes deprimidos, con inhibición psicomotora y con niveles disminuidos de HMPG responderían mejor a la desipramina y a la imipramina, en tanto que aquellos con depresión ansiosa y con niveles normales de HMPG mejorarían con el uso de la amitriptilina, pero los resultados clínicos no sustentaron esta hipótesis.
Estudios posteriores utilizaron(6,7) los niveles séricos del HMPG, para precisar el empleo de imipramina o amitriptilina, en pacientes deprimidos con niveles disminuidos, en tanto que usaron antidepresivos inhibidores selectivos de la recaptación de serotonina (ISRS) en aquellos con cifras normales. Estudios que presentan fallas metodológicas e impiden generalizar los resultados obtenidos.
Una investigación adelantada por Garvey y Tuason(8) con 16 pacientes deprimidos, con historia familiar de trastornos depresivos, encontró que los niveles de HMPG no se modificaron después de seis semanas de tratamiento. Este resultado permitió concluir que la alteración del metabolismo de la nora-drenalina y los consiguientes niveles bajos del HMPG se deben a un mecanismo bioquímico adaptativo existente en los pacientes deprimidos, y que por lo tanto no se puede considerar como un predictor de la respuesta farmacológica.
Datos tan disimiles en las concentraciones urinarias y plasmáticas del HMPG permiten concluir a los investigadores, que este metabolito, por si solo, no es un buen indicador del metabolismo de la noradrenalina ni un predictor de la respuesta farmacológica.
NORADRENALINA Y RECEPTORES
Los receptores noradrenérgicos son de dos clases: alfa y beta. Los receptores alfa-1 son excitadores y están localizados en el cerebro, las fibras musculares lisas vasculares e intestinales, y el músculo cardíaco. Su estimulación produce aumento del calcio intracelular y activación de los sistemas del fosfatidilinositol y de la proteinkinasa C.
Los receptores alfa-2 son inhibidores y se encuentran en el cerebro, las fibras musculares lisas vasculares e intestinales, la terminal nerviosa y las plaquetas, donde parece estar relacionado con el fenómeno de agregación plaquetaria. Su acción origina disminución de la ade-nil ciclasa e inhibición de los canales del calcio. Este se corríporta como un autorreceptor, es decir, es el responsable de la información de la retroalimentación en la producción de noradrenalina: su excitación produce disminución de la liberación del neurotransmisor, en tanto que al ser bloqueado se produce mayor liberación por parte de las neuronas noradrenérgicas.
Los betarreceptores son denominados beta-1, beta-2 y beta-3 y su acción excitadora o inhibitoria utiliza la vía de la adenilciclasa. Los receptores beta-1, están localizados a nivel cardíaco y son excitadores. Los beta-2 también tienen acción excitadora y están localizados en el músculo liso, el músculo estriado, el hígado y los linfocitos. Los receptores beta-3 se localizan en el tejido graso.
Los antidepresivos noradrenérgicos actúan sobre los receptores beta del cerebro y de los linfocitos produciendo una disminución de la respuesta del receptor.
La noradrenalina activa los receptores beta-adrenérgicos responsables de la activación de la vía del monofosfato cíclico de adenosina (AMPc), de los segundos y terceros mensajeros de la trasducción intracelular y del sistema regulador de los factores neurotróficos de la neurona.
La primera acción ocurre mediante fosforilación y regulación de la proteinkinasa del AMPc, de la fosfoli-pasa C, del fosfoinositol-3 del receptor post-sináptico y las proteinkinasas dependientes del calcio(9).
Los receptores alfa-2 actúan como autorreceptores en la terminal no radrenérgica y su afinidad aumenta en los estados depresivos, lo cual sugiere una alteración en la liberación de noradrenalina. Además, se ha encontrado una respuesta plana del AMPc al estimular el adrenorreceptor alfa-2, lo cual hace pensar en la existencia de una alteración a nivel del receptor (10).
El receptor N-Metil-D Aspartato (NMDA) juega un papel importante en la modulación de la transmisión noradrenérgica en el SNC. Los aminoácidos que excitan el receptor NMDA producen aumento en la estimulación de liberación de noradrenalina en el hipocampo(11).
INTERACCIONES ENTRE NORADRENALINA Y SEROTONINA
Varios hechos clínicos y neuroanatómicos hacen suponer la existencia de un sistema de retroalimentación entre noradrenalina y serotonina:
- Niveles elevados de noradrenalina activan el autorreceptor alfa-1 en la neurona serotoninérgica, aumentando la tasa de disparo y liberación de serotonina.
- Las fibras noradrenérgicas inervan los núcleos del rafe donde están situadas las neuronas serotoni-nérgicas. A su vez, la serotonina puede inhibir la actividad del locus ceruleus , especialmente la relacionada con/los estímulos dolorosos, mediantje la acción sobre los receptores 5-HTla y el 5-HT2a, utilizando la vía de los aminoácidos excitadores, como el glutamato (12). Este mecanismo explicaría la eficacia clínica de la amitriptilina en el tratamiento del dolor crónico de tipo neuropático.
- Algunas funciones noradrenérgicas como la regulación del sueño, la ansiedad y la irritabilidad son compartidas con los mecanismos serotoninérgicos, como se esquematizó en la figura 1.
- Las crisis del trastorno de pánico parecen ser originadas por una desincronización del locus ceru-leus; razón por la cual, los antidepresivos noradrenérgicos serían los fármacos indicados para su tratamiento. Sin embargo, la experiencia clínica ha demostrado que sólo la imipramina y la clomipramina, que poseen acción noradrenérgica y serotoninérgica, son los únicos antidepresivos que han demostrado ser eficaces. Por otra parte, se han obtenido excelentes resultados terapéuticos al utilizar los ISRS y los inhibidores de la mono-amino-oxi-dasa (MAOS), fármacos de un marcado perfil serotoninérgico.
- El pindolol un betabloquedor noradrenérgico ha demostrado poseer acción serotoninérgica. Algunos investigadores, lo han empleado junto con los ISRS, tipo fluoxetina, obteniendo una respuesta antidepresiva más temprana que disminuye la duración del período de latencia del antidepresivo.
NORADRENALINA Y DEPRESIÓN
En los estados depresivos se ha observado desincronización de las descargas de noradrenalina del locus ceruleus que podrían explicar las anormalidades de sus funciones a nivel central y periférico.
En el estrés crónico, el hipotálamo secreta hormona liberadora de corticotropina (CRH) que al actuar sobre el locus ceruleus incrementa la síntesis de noradrenalina y al mismo tiempo disminuye la utilización de los cambios adaptativos del individuo, lo cual se traduce en incremento en la concentración del neurotrasmisor. Este modelo de estrés crónico se emplea, por analogía, como modelo para la enfermedad depresiva y permite evaluar los cambios en el sistema noradrenérgico: reducción de la densidad de los beta-adrenorreceptores, aumento en la densidad de los receptores alfa-1 y alfa-2 y disminución de la respuesta del AMPc(13).
Los cambios en los adrenoreceptores conforman una función adap-tativa (plasticidad neuronal) cuyo objetivo es disminuir el desarrollo de la enfermedad depresiva, pero resultan insuficientes para impedir la aparición de los síntomas depresivos (Figura 4).
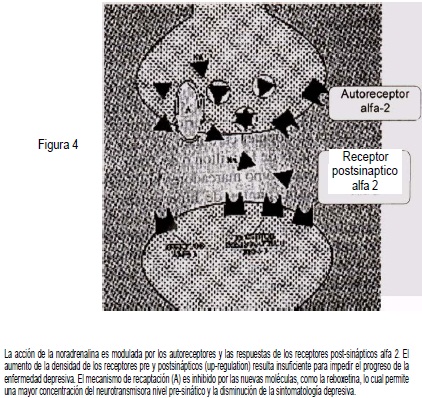
Esta afirmación ha sido corroborada por las investigaciones de Leo-nard(13) quien creó un modelo animal de la depresión., Al extirpar los bulbos olfatorios de la rata, se producen cambios en la función de la amígdala, en donde disminuyen las concentraciones de noradrenalina; disminución que origina aumento de la densidad de los adrenorreceptores alfa-1 y alfa-2, modificaciones similares a las observadas en los pacientes deprimidos.
Los cambios adaptativos post-bulbectomía olfatoria desaparecen al cabo de algunas semanas de tratamiento con reboxetina (inhibidor selectivo de la recaptación de la noradrenalina).
Diferentes hechos sugieren que existe un déficit en la actividad noradrenérgica responsable de la aparición de los síntomas depresivos:
- Niveles urinarios de MHPG frecuentemente disminuidos en los pacientes con depresión endógena(5,6,7,14,15).
- Menores concentraciones urinarias de HMPG en los pacientes con depresión bipolar que los pacientes con depresión monopolar(14/15).
- Tanto los antidepresivos como la terapia electroconvulsiva (TEC) producen down-regulation de los beta-adrenorreceptores, acción que es aceptada por varios autores como un marcador bioquímico de la eficacia antidepresiva(12-16).
- Los siguientes hallazgos en pacientes suicidas:
- *Aumento de los receptores de enlace beta-1-adrenoreceptores que en los grupos control(17).
- *Incremento de la densidad y de la afinidad de los adrenoreceptores alfa-2, lo cual sugiere que la depresión puede estar relacionada con la hipersensivilidad de estos receptores(18).
- *Disminución del número de células noradrenérgicas del locus ceru-leus y aumento en las concentraciones de tirosina hidroxilasa(19). Hallazgo no validado por otros autores que han encontrado disminución la enzima(4-13).
- *Disminución de los sitios de enlace de la hormona liberadora de corticotropina en la corteza frontal(20).
- Disminución del número de beta-adrenorreceptores en los linfocitos de los pacientes deprimidos y un aumento en la respuesta de los beta-adrenoreceptores posterior a la terapia electroconvulsiva(21).
- Estudios clínicos que sugieren que existe una hipersensibilidad de los adrenorreceptores alfa-2 en la enfermedad depresiva(22).
- Reportes de disminución de la agregación plaquetaria inducida por la noradrenalina (vía adenil ci-clasa y AMPc) en los pacientes deprimidos, que sugiere una disminución de los alfa-2 adrenoreceptores plaquetarios(23).
- Evidencia de que estrés crónico y la reserpina aumentan la tasa de disparo (firing) del locus ceruleus y aumentan la secreción de tirosina hidroxilasa, que disminuye con el tratamiento antidepresivo prolongado con imipramina. Los estudios sugieren que en la depresión existe un trastorno en la actividad de la tirosina hidroxilasa y que los antidepresivos actuarían a nivel de la expresión genética de la tirosina hidroxilasa, produciendo un fenómeno de down-regulation* en la biosíntesis de noradrenalina.
- El hallazgo en pacientes deprimidos de disminución de la proteína transportadora de noradrenalina en el locus ceruleus, como un mecanismo compensatorio del tipo down-regulation debida a la biodisponi-bilidad disminuida de noradrenalina(24).
- Los alfa-2-adrenorreceptores controlan la secreción de la hormona liberadora de la hormona del crecimiento (GHRF). Se ha observado una respuesta plana de la liberación de la hormona del crecimiento al administrar clonidina en los pacientes deprimidos, respuesta que persiste aun cuando el paciente se haya recuperado, razón por la cual algunos autores consideran esta alteración hormonal, como un marcador biológico de predisposición a la enfermedad depresiva(22-25).
- Elevación en la secreción de la CRH y de su proteína trasportado-ra en pacientes deprimidos e inclusive disminución de los sitios de enlace de la CRH en la corteza frontal de los suicidas, comparados con el grupo control(20).
- La hormona tiroidea por aumento de la actividad del receptor beta-adrenérgico promueve la acción de la noradrenalina, razón por la cual disminuye la liberación nocturna de la hormona tiroestimulante (TSH) y se presenta uan respuesta plana al estimular su producción con TRH. La respuesta plana del test TRH-TSH se considera como un factor de riesgo para padecer depresión y un marcador biológico de cronicidad.
- La síntesis y secreción nocturna de melatonina es controlada por la transmisión noradrenérgica de la glándula pineal que convierte la serotonina en melatonina. Los antidepresivos aumentan la actividad noradrenérgica que disminuye la producción de melatonina y explicaría, en parte, las alteraciones del sueño que presentan los pacientes deprimidos.
- La terapia electroconvulsiva aumenta la liberación de noradrenalina pero algunos autores han encontrado disminución del neurotrasmisor en pacientes que se habían recuperado del cuadro melancólico.
* Cuando existe disminución de la concentración del neurotransmisor se produce un incremento en el número de receptores pre y postsinápticos. Este fenómeno es llamado up regulation. Cuando se recuperan los niveles del neurotransmisor disminuyen el número y la sensibilidad de los receptores (down regulation) se recupera la función sináptica.
En resumen, en el episodio depresivo se presenta disminución de las células noradrenérgicas y aumento de la tirosina hidroxilasa a nivel del locus ceruleus, que origina una disminución de la biodisponibilidad de noradrenalina.
Las menores concentraciones de noradrenalina producen aumento de la producción de CRH y corticotropina (ACTH) que originan un aumento de las cifras de cortisol, disminución de la respuesta de la hormona del crecimiento a la hipoglicemia, disminución de la producción de la hormona luteinizante, aumento de los receptores alfa-2 en las plaquetas, en tanto que los receptores beta-2 de los linfocitos permanecen inalterados. Por último, a nivel urinario se encuentra una mayor excreción de noradrenalina y HMPG.
Estos cambios moleculares son compartidos por los pacientes deprimidos y por quienes padecen anorexia nerviosa, lo cual explica la comorbilidad de ambos cuadros clínicos. En los anoréxicos las alteraciones biomoleculares parecen ser más graves, si se tiene en cuenta su mortalidad elevada y su escasa respuesta al tratamiento antidepresivo(3-27).
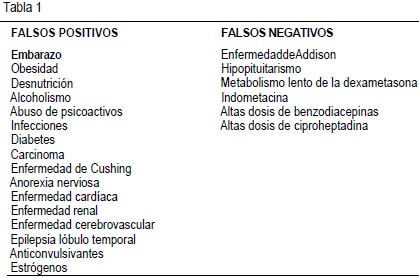
Se ha tratado de utilizar como marcador biológico la hipercortisolemia que se presenta en el episodio depresivo, mediante su inhibición con dexametasona. Infortundamente el test de la supresión de la dexametasona no es específico y diferentes entidades muestran resultados falsos positivos y falsos negativos (Tabla 1). Hoy se acepta que los pacientes que no normalizan los resultados del test de la dexametasona al terminar su tratamiento antidepresivo tienen una mayor tendencia a presentar recaídas clínicas(28).
NORADRENALINA Y ANTIDEPRESIVOS
Los fármacos antidepresivos parecen actuar mediante adaptaciones de la trasducción intracelular de señales y la regulación de la expresión genética que son los mecanismos responsables de la plasticidad celular.
Los antidepresivos producen down-regulation de la densidad de los receptores beta-adrenérgicos y de la estimulación del AMPc, como respuesta al incremento de la concentración de noradrenalina a nivel sináptico.
El tratamiento con antidepresivos noradrenérgicos induce, al parecer, traslocación de la proteinkinasa dependiente del AMPc a nivel de la corteza cerebral e influyen sobre la expresión del receptor beta-adrenérgico del RNA mensajero, que regula la trascripción de este receptor, y de las expresiones de las subu-nidades de la proteína G y de la adenilciclasa cerebrales(9-29).
La desensibilización del receptor beta-adrenérgico y la disminución de la producción del AMPc requieren cerca de dos semanas (período de latencia de los antidepresivos). No es la acción sobre el receptor sino los cambios bioquímicos y fisiológicos a nivel de los componentes post-sinápticos e intracelulares, tales como la activación del sistema de la adenilciclasa y las proteínas Gs, los responsables de la recuperación de la actividad intrínseca neuronal y de la acción antidepresiva(29).
Los autoreceptores del soma, las dendritas y el axón terminal son alfa-2 pero los receptores post-sinápticos son de tres tipos: alfa-2a, alfa-2b y alfa-2c. El enantiómero (+) de la mirtazapina bloquea con una mayor potencia los autoreceptores (alfa-adrenorreceptores) que los receptores post-sinápticos, aumentando así el disparo (firing) y liberación de noradrenalina por parte de la neuronas noradrenérgicas, cuya concentración extracelular corresponde a los niveles del ácido dihidro-fenil-acético (DOPAC) a nivel del hipocampo, obtenidos por microdiálisis(12).
En síntesis, la acción terapéutica de los antidepresivos noradrenérgicos se realiza mediante un mecanismo de down-regulation de los beta-adrenoreceptores y una sensibilización (up-regulation) de los alfa-2-adreno-receptores.
En la vía de la noradrenalina actuarían los siguientes antidepresivos que facilitan la acción noradrenérgica cerebral mediante mecanismos como la inhibición de la recaptación, inhibición del metabolismo, estimulación de la liberación del neurotransmisor o modulación de la función del receptor post-sinaáptico, el alfa-2-adrenoreceptor(30).
- Inhibidores de la recaptación:
- Inhibidores selectivos: reboxetina, teniloxacina.
- Antidepresivos heterocíclicos: clomipramina, imipramina, amitriptilina, maprotilina, trimeprimina, nortriptilina, doxepina, desipramina.
- Moduladores adrenérgicos: amisulpride, bupropion.
- Doble recaptación de serotonina y noradrenalina: venlafaxina y diluxetina.
- Atípicos: nefazodone
- Inhibidores de la MAO:
- No reversibles o clásicos: fenelci-na, tranilcipromina, isocarbaxida.
- Reversibles: moclobemida (IMAO-A), selegilina (IMAO-B).
- Antagonistas del receptor alfa-2: mianserina, mirtazapina
La presencia de noradrenalina es imprescindible para la acción terapéutica de los antidepresivos noradrenérgicos. Igual sucede con la serotonina que es indispensable para la acción de los ISRS y los IMAOS. Delgado y colaboradores administraron a pacientes con mejoría estable después del tratamiento antidepresivo una dieta baja en triptofano, el aminoácido precursor de la serotonina, y rica en aminoácidos que competían con la protei-na trasportadora del triptofano. Al cabo de dos semanas recayeron en la depresión el 70% de los pacientes que habían mejorado con ISRS o IMAOS y el 20% de los pacientes tratados con desipramina, nortriptilina y bupropion. Al administrar alfa-metil-paratirosina, un inhibidor de la noradrenalina, los pacientes que estaban recibiendo antidepresivos noradrenérgicos presentaron síntomas de recaída clínica(31).
Estos resultados permiten concluir que la clave del efecto terapéutico de los antidepresivos es la recaptura de la noradrenalina o la serotonina y no su efecto a nivel de los receptores pre y post-sinápticos.
Los antidepresivos tricíclicos son los fármacos noradrenérgicos más usados actualmente y uno de ellos, la imipramina, es considerado por los investigadores como el «patrón de oro» de los antidepresivos. Workman y Short hicieron un estudio de meta-análisis de los ensayos clínicos con bupropion, fluoxetina, trazodone e irniprarnina comparados con placebo y publicados en el período 1980-1990. La muestra correspondió a 2.090 pacientes en los cuales se pudo comprobar que ningún antidepresivo demostró ser mejor que la imipramina (32).
El trazodone, la trimeprimina y la mirtazapina facilitan la trasmisión noradrenérgica al aumentar la tasa de disparo del locus ceruleus. La mirtazapina, además, bloquea los adrenorreceptores alfa-2, por acción de su enantiómero (+) aumentando la concentración de noradrenalina en la hendidura sináptica.
El hidroxibupropion, metabolito del bupropion, inhibe la tasa de disparo de las neuronas noradre-nérgicas del locus ceruleus, cuya concentración inhibe la tasa de disparo de las neuronas dopaminérgicas del cerebro medio(12).
Los inhibidores selectivos de la re-captación de noradrenalina (NARI) son fármacos capaces de corregir la disfunción noradrenérgica sin ocasionar los efectos indeseables de los tricíclicos al no actuar sobre los receptores alfa-adrenérgicos e hista-minérgicos.
La reboxetina y la teniloxacina inhiben la recaptación de la noradrenalina, disminuyen la sensibilidad de los adrenoreceptores alfa-2, modifican la actividad del sistema de fosforÜación neuronal dependiente del sistema calcio-calmodulina y su administración durante un período prolongado originan plasticidad neuronal capaz de contrarrestar la disfunción noradrenérgica(33,34).
Los estudios clínicos han demostrado que la reboxetina mejora hacia la segunda semana de tratamiento el afecto deprimido, la desesperanza y la anhedonia, y aumenta la motivación , el interés por el trabajo y las actividades sociales(35,36,37). Este hecho clínico permite suponer que los NARI serían los antidepresivos más adecuados para tratar los pacientes melancólicos con marcado retardo psicomotriz, en tanto que los ISRS serían los antidepresivos para preferir en el tratamiento de la depresión ansiosa(38).
Las nuevas investigaciones sobre la actividad noradrenérgica y sus implicaciones en la etiología del trastorno depresivo han reafirmado la hipótesis de una etiología poligénica y multifactorial de los cuadros depresivos, en la cual varios neuro-transmisores (noradrenalina, serotonina, dopamina, GABA, colecistokinina) y diferentes cambios moleculares a nivel neuronal (hipersensibilidad de receptores, modificación en los autorreceptores, cambios en los segundos y terceros mensajeros) son responsables de la aparición y cronicidad de los síntomas depresivos.
La evaluación clínica precoz de los síntomas es indispensable para evitar el menoscabo en la calidad de vida del paciente.
Ante la ausencia de un antidepresivo ideal, las nuevas moléculas inhibidoras selectivas de la recaptación de noradrenalina poseen a su favor una buena eficacia antidepresiva, un menor número de efectos secundarios y se muestran promisorias al lograr que el paciente deprimido se reintegre en una forma temprana a sus actividades cotidianas.
REFERENCIAS
1. Schildkraut JJ: The catecholamine hypothesis of affective disorders. A review of supporting evidence. Am J Psychiatry 1965; 122:509-522. [ Links ]
2. Bunney WE Jr, Davis DM : Norepinephrine in depressive reactions. Arch Gen Psychiatry 1965; 13(6): 483-494. [ Links ]
3. Pirke KM. Central and peripheral noradrenalln regulation in eating dlsoraers. Psychiatry Res 1996; 62(1):43-49. [ Links ]
4. Leonard BE. Fundamentáis of Psychopharmacology. John Wiley & sons (ed). Second Edition. Chichester, England 1997. [ Links ]
5. Maas JW, Fawcet J, DEKIRMENIAN H. 3-methoxy-4-hydroxy-phenylglicol (MHPG) excretion in depressive states. Arch Gen Psychiatry 1968; 19:129-134. [ Links ]
6. Beckman H, Goodwin FK. Antidepressant response to TCAs and urinary MHPG in unipolar patients. Arch Gen Psychiatry 1968; 19:129-134. [ Links ]
7. Schatzberg AF, Samson JA, Bloomingdale KL, Orsulak PJ, Gerson B, Kisukapp, Colé Jo, Shlidkraut JJ. Towards a biochemical classification of depressive disorders. X. Urinary catecholamines, their metabolltes and D-type scores in subgroups of depressive disorders. Arch Gen Psychiat 1989; 46:260-268. [ Links ]
8. Garvey MJ.Tuason VB. Do low levels of MHPG in depressive spectrum patients normalize after successful treatment?. Neuropsychobiology 1996; 34(4):188-191. [ Links ]
9. Nestler EJ, Duman RS. Relevance of intracellular signal trasduction pathways to Psychiatry. In: American Psychiatric Press Annual Review. Volume 15. American Psychiatric Press Inc. Washignton, DC. 1996;279-308. [ Links ]
10. Garcia-Seville JA, Zis AP, Selnic JS, Smith CB. Platelet alpha 2-adrenergic receptor in major depressive disorder. Arch Gen Psychiatry 1981; 38:1327-1333. [ Links ]
11. Fink K.Schultheiss R, Gothert M. Stimulatlon of noradrenallne release in human cerebral cortex mediated by N-methyl-D-aspartate (NMDA) and non-NMDA receptors. Br J Pharmacol 1992;106:67-72. [ Links ]
12. Frazer A. Pharmacology of antidepressants. J Clin Psychopharmacol 1997;17(2) (suppl): 3-18. [ Links ]
13. Leonard BE. Noradrenaline in basic models of depression. Eur Neur Psychopharmacol 1997, 7(suppl 1): 11-16. [ Links ]
14. Muscettola G, Potter WZ, Pickard D, Goodwing FK. Urinary 3-methox¡-4-hydroxy-phenyl-glicol and major affective disorders. Arch Gen Psychiatry 1984; 41:337-342. [ Links ]
15. Schildkraut JJ, Orsulak PJ, Labrie Ra, Schatzberg AF, Gudeman JE, Colé OJ et al. Towards a biochemical classification of depressive disorders. II. Application of multivariate discriminant function analysis to data on urinary cathecolamines and metabolites. Arch Gen Psychiat 1978; 35:1436-1439. [ Links ]
16. Werstiuk ES, Coote M, Griffith L, Shannon H, Steiner M. Effects of electroconvulsive therapy on peripheral adrenoreceptors, plasma noradrenaline, MHPG and cortisol in depressed patients. Br J Psychiatry 1996; 169(6):758-765. [ Links ]
17. Mann JJ, Brown RR, Halper JR, Sweeney J. Reduced sensitivity of lymphocyte beta-adrenergic receptors in patientes with endogenous depression and psychomotor agitation. New Engl J Med 1985; 313:715-720. [ Links ]
18. Meana JJ, Barturen F, Garcia-Sevilla JA. Alpha2-adrenoceptors in the brain of suicide victims: increased receptor density associated with major depression. Biol Psychiatry 1992;31:471-490. [ Links ]
19. Arango V, Underwood MD, Mann JJ. Biologic alterations in the brainstem of suicides. Psychiatr Clin North Am 1997; 20(3):581-593. [ Links ]
20. Nemeroff LB, Owenas MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropine releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psych 1988; 45:577-579. [ Links ]
21. Maan JJ, Mahler JC, Wilner PJ, Halper JR Brown RR Johnson KS et-al. Normalization of blunted lymphocyte beta-adrenergic resposivity ib melancholic inpatients by a course of electroconvulsive therapy. Arch Gen Psychiatry 1990; 47:461-464. [ Links ]
22. Charney DS, Heninger GR, Sternberg D, Redmond DE, Leckman JF et al. Presinaptic adrenergic receptor sensitivity in depression. The effect of long-term desipramine treatment. Arch Gen Psychiat 1981; 38:1334-40. [ Links ]
23. Nunget DF, Diñan TG, Leonard BE. Alteration by a plasma factor(s) platelet aggregation in unmedicated unipolar depressed patients. J Affect Dis 1994; 31:61-66. [ Links ]
24. Klimek V, Stockmeier C, Overholser J, Metzer KK, Kalka S, Dilley G, et al. Reduced levels of norepinephrine transports in the locus ceruleus in major depression. J Neurosc 1997; 1 7(21 ):8451-8458. [ Links ]
25. Mitchell PB, Bearn JA, Corn TH, Checkley SA. Growth hormone response to clonidine after recovery in patients with endogenous depression. Br J Psychiatry 1988; 152:34-38. [ Links ]
26. Kelly CB, Cooper SJ. Plasma noradrenaline response to electroconvulsive therapy in depressive illness. Br J Psychiatry 1997; 171:182-186. [ Links ]
27. Beumont PJV The nuerobiology of eating behaviour and weight control. In: Neuroblology in the treatment of eating disorders. Hoek HW, Treasure JL, Katzman M (editors). John Wiley & sons, Chichester, England. 1998. [ Links ]
28. Janicak PG, Davis JM, Preskorn SH, Ayd FJ. Principies and practice of Psychopharmacotherapy. 2nd Edition. Williams & Wilkins (ed) Baltimore, 1997. [ Links ]
29. Chen J, Rasenlck MM. Chronic antidepressant treatment facilítales G protein activation of adenllyl cyclase without altering G protein content. J Pharm ExpTher 1995; 275:509-517. [ Links ]
30. Tellez J. Antidepresivos en la práctica médica. Nuevo Milenio Editores. Bogotá, 1998. [ Links ]
31. Delgado PL, Miller HL, Salomón RM, Licinio J, Henninger GR. Monoamines and the mecanism of antidepressant action: effects of catecholamlne depletion on mood of patients treated with antidepressants. Psychopharmacol Bull 1993; 29:389-396. [ Links ]
32. Workman EA, Short DD. Atypical antidepressants versus imipramine in the treatment of major depression: a meta-analysls. J Clin Psychiatry 1993; 54:5-12. [ Links ]
33. Brunello N, Ragacni G. Rationale for the development of noradrenaline reuptake inhibitors. Hum Psychopharmacol 1998; 13(supl 1): S13-S20. [ Links ]
34. Szabadi E, Bradshaw CM, Boston PF, Langley RW. The human pharmacology of reboxetine. Hum Psychopharmacol 1998; 13(supl 1):S3-S12. [ Links ]
35. Dubini A, Bosc M, Polin V. Do noradrenaline and serotonin differentially affect social motivation and behaviour?. Eur Neuropsychopharmacol 1997; 7(suppl 1):S49-S56. [ Links ]
36. Healy D, MC Monagle T. The enhancement of social functioning as a therapeutic principle in the management of depression. J Psychopharmacol 1997; 11(suppl 1):S25-S31. [ Links ]
37. Hindmarch I. Effects of antidepressants on cognitive and psychomotor junction: the lack of effects of reboxetine. Hum Psychopharmacol 1998; 13(suppl 1):S21-S28. [ Links ]
38. Diñan TG. Noradrenergic and serotonergic abnormalities in depression: stress-induced disfunction?. J Clin Psychiatry 1996;57(suppl 4):14-18. [ Links ]

