










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.29 no.4 Bogotá Oct./Dec. 2000
ARTICULO ORIGINAL
DEPRESIÓN POSTISQUEMIA CEREBRAL UNA APROXIMACIÓN FISIOPATOLÓGICA Y CLÍNICA
POSTSTROKE DEPRESSION A PHYSIOPATHOLOGICAL AND CLINICAL APPROXIMATION
CÉSAR A. ARANGO D* HERNÁN J. PIMIENTA J. ** MARTHA I. ESCOBAR B. ***.
* Médico Psiquiatra, aspirante a Doctorado en Ciencia Biomédicas Universidad del Valle. Email: arangodavila@telesat.coiii.co
** Profesor Titular y Director del Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
*** Profesora Titular, Facultad de Salud, Universidad del Valle - Investigadora del Centro de Estudios Cerebrales
La depresión y otras manifestaciones emocionales pueden ser consecuencia de lesiones isquémicas cerebrales focales. La particularidad clínica, la relación con la localización de la lesión, la evolución diferencial según el hemisferio afectado y los estudios imagenológicos funcionales permiten reconocer el papel de los factores biológicos en esta patología. Datos de modelos de isquemia experimental focal, en las que se establecen cambios en los receptores de benzodiacepinas, en proteínas asociadas a los microtúbulos (MAP2) y en la proteína acídica fibrilar glial (GFAP) en sectores alejados del foco isquémico y relacionados con las estructuras límbicas, sustancia negra o sustancia gris periacueductal, permiten formular una hipótesis fisiopatológica que implica los circuitos cortícoestriato-tálamo-corticales y los diferentes sistemas de modulación provenientes del tallo cerebral como: serotonina, norepinefrina, dopamina y acetilcolina. Se presenta la hipótesis del posible papel neuroprotector de los antidepresivos.
Palabras clave: Isquemia; Depresión.
Depression and other emotional manifestatíon may emerge after a focal cerebral ischemic injury. The conspicuous clinical character, its relationship with the site of the lesión, the dissimilar evolutíon depending on the affected hemisphere and the imagenological studies support the concept that biological factors are associated with this pathology. Several lines of evidence based on experimental models of focal cerebral ischemia have demonstra ted exofocal changes in differentneuronal markers such us benzodiazepine receptors, glial fibrillary acidic protein and microtubule-associated -protein 2, that may contribute to the expression of these affective symptoms. In our study we observed that these changes involved sustantia nigra, periaqueductal gray matter and raphe nuclei, áreas connected with the infarcted área. A physiopathological hypothesis is formulated considering the relationship between corticostriato- thalamic circuits and their main system of regulation i.e. dopamine, serotonin, norepinephrine and acetilcholine. Some aspects of the possible role of antidepressants as neuroprotectors are also discussed.
Key words: Ischemia; Depression.
INTRODUCCIÓN
En los últimos años la prevalencia de la isquemia cerebral ha disminuido, sin embargo, todavía ocupa los primeros lugares de morbimortalidad en el mundo (1,2).
La depresión representa la principal secuela psiquiátrica de las lesiones vasculares focales (3,8). Otras alteraciones emocionales como la manía, el trastorno bipolar, los trastornos de ansiedad, también se pueden presentar aunque no con la frecuencia de la depresión(9,13).
Diferentes estudios demuestran la relación existente entre isquemia cerebral y manifestaciones depresivas, las cuales se presentan entre 12% y 65% de los pacientes (Tabla 1). Depresión que disminuye significativamente la calidad de vida de las personas en los años posteriores a la lesión (14,16). Algunas evidencias indican que estas manifestaciones psicopatológicas se deben a alteraciones orgánicas del encéfalo más que a respuestas adaptativas del individuo (17,19).

En el presente artículo se revisan los aspectos clínicos de la depresión posterior a una lesión isquémica cerebral focal, se especifican las características clínicas, los aspectos nosológicos, el desempeño en las actividades diarias y la relación entre la localización de las lesión y las manifestaciones depresivas. Se propone una hipótesis fisiopatológica apoyada en resultados experimentales y se intenta una aproximación farmacológica.
ASPECTOS NOSOLÓGICOS
La CIE10 (30) caracteriza un grupo de entidades clínicas denominadas "Trastornos Mentales debidos a lesión o Disfunción Cerebral o a Enfermedad Somática", entre los cuales incluye la enfermedad vascular. Para fundamentar tal clasificación considera cuatro criterios:
1. Evidencia de una enfermedad, lesión o disfunción cerebral acompañada de un trastorno del estado de ánimo.
2. Coincidencia en el tiempo entre el desarrollo de la enfermedad subyacente y el inicio del síndrome psicopatológico.
3. Remisión del trastorno mental cuando mejore la presunta causa subyacente.
4. Ausencia de otra etiología que explique el síndrome psicopatológjco.
Los dos primeros criterios son suficientes para justificar un diagnóstico provisional, mientras que la confluencia de los cuatro nos aproxima a un diagnóstico más certero.
Para el DSMIV(31) la depresión postisquemia cerebral corresponde al capítulo de "Trastornos del estado de ánimo debido a enfermedad médica", caso en que se consideran los síntomas mentales como una consecuencia directa de la enfermedad orgánica. Los siguientes son algunos de los criterios para establecer el diagnóstico según el DSM-IV: en primer lugar, se debe encontrar una asociación temporal entre el inicio, la exacerbación o la remisión de la condición médica general y los síntomas emocionales; en segundo lugar, algunas de las características clínicas del trastorno afectivo son atípicas con relación al cuadro afectivo primario, por ejemplo, edad de inicio, curso o historia familiar o personal del mismo; por último, se debe descartar la posibilidad de que el trastorno emocional corresponda a un trastorno afectivo primario, a un trastorno afectivo inducido por sustancias o a un trastorno del ajuste.
ASPECTOS CLÍNICOS
Aunque algunos autores no encuentran diferencias entre la fenomenología clínica de la depresión mayor y la depresión postisquemia cerebral (24,32) otros hallan manifestaciones clínicas más frecuentes en esta última, que en algunos casos son atípicas al compararlas con el trastorno depresivo mayor. Estas son:
• Irritabilidad (15,33)
• Egocentrismo (15)
• Llanto patológico(34)
• Risa patológica.(34)
• Lentitud o inhibición motora (7)
• Síntomas vegetativos (35)
• Ideación suicida, especialmente en pacientes con lesiones en el hemisferio izquierdo (6)
Posterior a una isquemia cerebral se pueden observar dos síndromes de presentación frecuente, clínicamente no muy bien definidos y cuya relación con la depresión no está claramente establecida: el emocionalismo o labilidad afectiva(33,34) y la apatía o indiferencia emocioné (35,36,37).
El emocionalismo es un estado en el cual ocurren cambios bruscos del estado de ánimo no relacionados con sucesos externos o como reacción a estímulos no significativos.
Se puede considerar que una manifestación más intensa de este síntoma es la denominada incontinencia afectiva (imposibilidad de reprimir las emociones de forma acentuada ante sucesos no relevantes). En los dos casos la presentación más frecuente es el llanto de aparición súbita e irrefrenable que cede en segundos para reactivarse más tarde.
La apatía o indiferencia emocional se define como carencia de sentimientos o interés por el medio con disminución de la capacidad de reacción ante estímulos externos. Es una manifestación no infrecuente de las lesiones isquémicas cerebrales, encontrándose hasta en el 23% de los pacientes y asociada a la mitad de los casos con depresión mayor (36). La apatía, como secuela de la isquemia cerebral, es más frecuente en pacientes de edad avanzada con compromiso cognitivo y limitación en sus actividades diarias W.
La presentación y evolución longitudinal de la depresión postisquemia cerebral se relaciona con las manifestaciones clínicas iniciales(33,34) con el hemisferio lesionado(38) y en un porcentaje elevado se puede asociar a factores como severidad de la isquemia, edad, sexo, antecedentes depresivos, etc. (Tabla 2). Se ha observado que después de tres años, la mitad de los pacientes que durante el primer año presentaron depresión continúan sintomáticos, especialmente aquellos que no muestran mejoría a pesar del tratamiento indicado (17).

La diferencia entre los pocos tratamientos antidepresivos prescritos y el elevado número de pacientes con depresión postisquemia cerebral que muestran los estudios, hace pensar que muchos casos no son reconocidos ni por los familiares ni por los médicos (1539). Las consecuencias afectivas y cognitivas son comúnmente olvidadas, limitándose la atención a la fase clínica inicial y a los procedimientos de fisiatría (20,37,39). La evaluación clínica de una isquemia cerebral usualmente se orienta a determinar el compromiso neurológico. El seguimiento se hace observando las características de la recuperación implementando estrategias neuroprotectoras y sesiones de rehabilitación. Los aspectos emocionales adquieren importancia sólo cuando las manifestaciones afectivas son muy evidentes o perturban la dinámica social del paciente y de sus cuidadores.
ACTIVIDADES DE LA VIDA DIARIA
Los pacientes con síntomas depresivos, que han presentado depresión postisquemia cerebral, tienen mayor perturbación de las actividades diarias que aquellos no deprimidos (42,43): al tercer mes requieren del apoyo de otros para el desarrollo de sus labores y al cabo de un año no recuperan su capacidad funcional (17,38). En los pacientes con apatía y depresión es muy importante el compromiso de las actividades diarias ya que con frecuencia persisten con esta limitación (37).
Hay pocos estudios que permitan evaluar verazmente la respuesta emocional de los pacientes a la fisioterapia o a las intervenciones psicosociales, la satisfacción reportada por algunos terapistas no necesariamente es compartida por los pacientes o por sus familiares (37,44) Frecuentemente los pacientes son criticados por sus familiares o cuidadores especialmente por que se tornan retraídos, irritables y egocéntricos (25); la función sexual se encuentra disminuida en casi todos los casos (34) al igual que las actividades sociales y recreacionales (14).
Según Angeleri y colaboradores(15) el 21 % de los pacientes que han sufrido un episodio isquémico focal regresan a su actividad laboral. De estos, sólo el 30%, es decir el 6% del total, pueden reasumir las funciones laborales previas a su compromiso neurológico. El resto se integran a actividades con niveles de complejidad inferior a las que desarrollaban previamente (33,43). De acuerdo con lo anterior, aproximadamente el 70% de los pacientes están de alguna manera impedidos, a lo cual se agregan los síntomas depresivos en una alta proporción.
LOCALIZACION DE LA LESION Y DEPRECION POSTISQUEMIA CEREBRAL
El primer estudio sistemático que relaciona al hemisferio lesionado con las manifestaciones depresivas fue realizado por Gainottí en 1972 (45), el cual reportó que los pacientes con lesión en el hemisferio izquierdo, especialmente aquellos con afasia, presentaban mayor tendencia a la depresión que los pacientes con lesiones en el hemisferio derecho. Desde entonces se han realizado muchos estudios para encontrar la relación entre la localización de la lesión isquémica focal y las manifestaciones emocionales (1,6,13,46,47)
En la mayoría de estos se halla correlación entre la lesión vascular focal del hemisferio izquierdo y la depresión, lo cual se acentúa cuando la lesión es más próxima al polo frontal (7,17,19,23,27,35,4) Además, se ha establecido que las lesiones de áreas posteriores de los hemisferios, por lo general, no desencadenan cuadros depresivos con la frecuencia en que los ocasionan las lesiones anteriores (33,49). Astrom y colaboradores (17) han informado que las lesiones localizadas en sectores anteriores del hemisferio izquierdo tienen una tendencia tres veces mayor de producir depresión al compararlas con las lesiones posteriores y 10 veces mayor con relación a las lesiones del hemisferio derecho.
Los datos mencionados se pueden correlacionar con algunos hallazgos imagenológicos funcionales en pacientes depresivos. En 1984 apareció el primer reporte de tomografía por emisión de positrones y depresión (50). Este estudio se realizó en 26 pacientes deprimidos en los cuales se observó hipometabolismo frontotemporal izquierdo. Desde entonces han aparecido otros estudios que evidencian una tendencia mayor al compromiso metabólico o del flujo sanguíneo cerebral del hemisferio izquierdo en su área anterior con relación al derecho en personas deprimidas (51).
El estudio más representativo que utilizó la tomografía por emisión de positrones en isquemia cerebral fue realizado por Mayberg en 1988 (52). Al aplicar un agonista 5HT2 a 57 pacientes con depresión postisquemia cerebral observó que la unión de este agonista a los receptores de serotonina en el hemisferio derecho tiende a incrementarse en las áreas no lesionadas de dicho hemisferio y no ocurre lo mismo cuando la lesión es en el hemisferio izquierdo. A partir de estos hallazgos concluyó que la severidad de la depresión guarda una relación inversa con la unión del agonista al receptor. Si tenemos en cuenta que el receptor 5HT2 ejerce un efecto básicamente excitador en la corteza cerebral(53), la disminución de la afinidad del receptor por el agonista o su disminución podría guardar relación con la depresión postisquemia cerebral. Con relación al hemisferio lesionado y la evolución longitudinal del cuadro depresivo, Astrom y colaboradores (17) observaron, en un estudio a tres años, una tendencia según la cual los pacientes con compromiso del hemisferio izquierdo presentan un trastorno depresivo mayor durante el primer año después de la lesión y una proporción menor de depresivos al cabo de tres años. En los pacientes con lesión en el hemisferio derecho la tendencia es contraria, es decir, al tercer año la proporción de depresivos es mayor que durante el primer año (Figura 1). Una investigación reciente de seguimiento (dos años) a pacientes con lesión isquémica cerebral del hemisferio izquierdo o derecho presenta resultados similares (38).
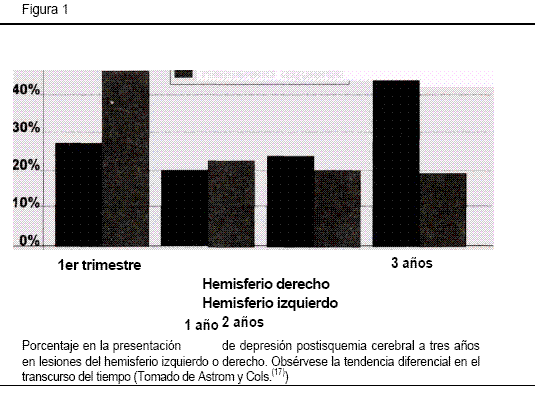
Adicionalmente se ha encontrado que lesiones de áreas anteriores de los ganglios básales izquierdos, pero no las lesiones talámicas, se asocian con depresión (7,23,41,54). Igualmente las lesiones isquémicas de los ganglios básales repercuten en el metabolismo cortical, interrumpiendo las conexiones corticosubcorticales generando a su vez alteraciones que implican los circuitos límbicos. Grasso y cois(55) encontraron que la perfusión de la corteza mesial temporal después de una lesión isquémica de los ganglios básales es significativamente menor en pacientes depresivos comparados con no depresivos. Este patrón se observa en pacientes deprimidos con daño en los ganglios básales derechos o izquierdos.
El tamaño de la lesión no logra demostrar relación con la depresión en la mayoría de los estudios (17,23,55). Sin embargo, se ha observado que pacientes con lesiones grandes en el hemisferio derecho presentan mayor tendencia a la depresión comparados con lesiones pequeñas en el mismo hemisferio; no ocurre así al comparar las lesiones grandes y pequeñas del hemisferio izquierdo (5).
Se han descrito algunos hallazgos imagenológicos asociados a la lesión focal que tienen relación con la presentación y especialmente con la persistencia de la depresión. Estos son atrofia cortical y subcortical (6,7,15,33) e hiperintensidad periventricular (resonancia magnética cerebral especialmente en pacientes seniles)(7).
FISIOPATOLOGÍA DE LOS CAMBIOS A DISTANCIA POSTERIORES A ISQUEMIA CEREBRAL
ASPECTOS GENERALES
Las lesiones ocasionadas por una isquemia cerebral han sido ampliamente estudiadas desde el punto de vista fisiopatológico. Las estrategias neuroprotectoras generalmente se dirigen a evitar la muerte de las neuronas, cuyos somas están localizados en las áreas de penumbra isquémica o en las de oligemia (56). De igual manera, la mayoría de los estudios experimentales que evalúan el efecto neuroprotector de un fármaco, restringen el análisis a determinar su repercusión sobre el tamaño de la lesión sin considerar los efectos sobre sectores alejados del foco isquémico (57,61).
La función del tejido nervioso se fundamenta en la interrelación electroquímica de unos sectores con otros a través de axones que se proyectan a distancia y localmente por medio de las complejas relaciones de las interneuronas. Por tanto, no es apropiado considerar una lesión cerebral que sólo produzca efectos localizados. En este caso, la extensa red de eferencias o proyecciones que se envían desde la región comprometida a zonas alejadas de la lesión isquémica van a desaparecer generando vacantes sinápticas exofocales. Paralelamente, neuronas cuyos cuerpos están distantes pero que proyectan sus axones al área lesionada, no van a poder interactuar sinápticamente con las células blanco del área necrótica ni podrán contactar neuronas fisiológicamente alteradas del área de penumbra o de oligemia. Así, el impacto sobre estas neuronas a corto y largo plazo no está claramente identificado.
También es importante considerar los efectos focales y exofocales de las lesiones isquémicas sobre las células gliales dada su íntima relación con las neuronas resultante de su papel regulador del microambiente iónico y la concentración de aminoácidos inhibitorios y excitatorios (62), estos últimos a través de transportadores de aminoácidos excitatorios de localización glial como el EAATl(GLAST) y el EAAT2 (GLT1) (63,64) La isquemia y otros factores que lesionan a las neuronas ocasionan, además, una respuesta glial que se generaliza a todo el hemisferio lesionado, pudiendo persistir por varias semanas y cuyas consecuencias a largo plazo en los sectores exofocales no está establecida (Figura 2).
DIASQUISIS
Von Monakov (65) en 1914 acuñó el término diasquisis para referirse a los cambios a distancia como consecuencia de una lesión cerebral. Específicamente sugirió que la disminución de las eferencias excitatorias del área lesionada se traduce en una reducción de la respuesta a los estímulos en sectores alejados del foco. Aunque el término diasquisis propuesto por Monakow se planteó originalmente en relación con la pérdida de la función en sectores distantes a un foco lesionado, con el transcurso del tiempo el concepto se ha ampliado e incluye la presencia de otros fenómenos como cambios eléctricos, del flujo sanguíneo y del metabolismo cerebral, neuroquímicos y neuroanatómicos micro y macroscópicos. Todas estas manifestaciones pueden describirse a distancia de la lesión isquémica, en las fases aguda, subaguda o crónica.
Los estudios en humanos sobre la actividad eléctrica, flujo sanguíneo y actividad metabólica durante las primeras 24 horas después de un infarto son pocos e inconsistentes. En varias especies: animales roedores, felinos y primates, sometidos a isquemia experimental, se han demostrado cambios electroencefalográficos, incrementos de la amplitud de los potenciales evocados somatosensoriales, disminución del metabolismo de la glucosa y disminución del flujo sanguíneo cerebral, especialmente en lesiones grandes (65,66)
En humanos, al igual que en los modelos experimentales, después de 24 horas de una lesión isquémica cerebral se encontró ensanchamiento de los potenciales evocados somatosensoriales que evolucionan en el tiempo dependiendo del sitio de la lesión: si se trata de un compromiso cortical el fenómeno es notable a partir de los 10 días postisquemia, en cambio si la lesión es subcortical los cambios eléctricos se instauran a partir del tercer día. En cuanto al flujo sanguíneo cerebral, entre los 7 y 10 días se observa una disminución en espejo en la corteza contralateral a la lesión. Este hecho cede gradualmente con el transcurso de los meses, sin embargo, en el hemisferio ipsilateral el fenómeno persiste, especialmente si la lesión involucra un volumen grande de tejido. El metabolismo de la glucosa y el consumo de oxígeno después de 24 horas postisquemia pueden estar disminuidos en el hemisferio contralateral y especular a la lesión (65,67).
CAMBIOS EXOFOCALES MICROESTRUCTURALES Y NEUROQUÍMICOS
En la actualidad hay un interés creciente por evaluar los cambios microestructurales y neuroquímicos exofocales posteriores a una lesión cerebral, pero hasta ahora esta posibilidad es más factible en modelos experimentales. Se considera que el esclarecimiento de estos fenómenos podría ofrecer alternativas terapéuticas para entidades como la depresión y otros fenómenos afectivos y cognitivos que se suceden en los pacientes que han sufrido isquemia cerebral.
Nuestro grupo, utilizando un modelo experimental de lesión cerebral dsquém ica (68,'69) por oclusión transitoria con sutura intraluminal de la arteria cerebral media en ratas adultas, se evaluaron los cambios en la expresión de receptores de benzodiazepinas y en el citoesqueleto neuronal. El análisis involucró regiones de penumbra, oligemia isquémica y regiones exofocales, es decir, estructuras no irrigadas por la arteria cerebral media ocluida, como la corteza cerebral contralateral, el hipocampo y en el tegmento mesencefálico, la sustancia negra compacta y reticulada, el área tegmental ventral, la sustancia gris periacueductal y grupos serotoninérgicos del rafe (70,71).
En aquellos animales en que se indujo una lesión frontoparietal y del caudado putamen se observó un incremento significativo del enlace para receptores de benzodiazepinas ipsilaterales a la lesión. El hecho es significativo en el giro dentado del hipocampo y en la sustancia negra, haciéndose notable en la primera semana postisquemia y progresando hacia la normalidad a partir de la tercera semana (72). Los sitios de enlace para las benzodiazepinas corresponden a unidades reguladoras del receptor GABAA cuya distribución es extensa a lo largo del encéfalo (73,74).
La proteína asociada a los microtúbulos (M AP2)(75) es esencial en las interacciones del citoesqueleto neuronal, confiere estabilidad a los microtúbulos y por ende es esencial para preservar la morfología neuronal. Esta proteína es particularmente sensible a la injuria traumática o a la isquemia: sufre cambios en su expresión(76,77,78) que se reconocen inmunohistoquímicamente con el uso de anticuerpos específicos. En experimentos realizados por nuestro grupo (71,72), encontramos que los cambios en esta proteína no se restringen a las zonas isquémicas, sino que compromete regiones como la corteza contralateral, la sustancia negra o la sustancia gris periacueductal (Figura 3).

También se han demostrado cambios en la inmunoreactividad a la proteína asociada a los microtúbulos MAP2 en trauma cerebral(79) y por el uso de agentes excitotóxicos como el ácido kaínico. A este respecto, Ayala y Escobar(80) encontraron fragmentación del citoesqueleto en la corteza ipsi y contralateral a la lesión. Nagasawa y colaboradores en un modelo de isquemia cerebral focal similar al utilizado por nosotros (81) encontraron cambios neuronales en la sustancia negra ipsilateral varios días después de la lesión isquémica cortical. Estos cambios incluyen reducción de la protein kinasa C a partir del tercer día (82,83), acumulación de calcio desde el tercer día hasta la cuarta semana (83), reducción de receptores de acetilcolina muscarínicos después del séptimo día(84) y reducción de receptores D1 (85) y A1 (86) después de 3 días.
Los datos anteriores indican que, más allá de las regiones comprometidas directamente por la lesión, ocurren adaptaciones que pueden subyacer al deterioro progresivo de las funciones cerebrales postlesión, entre estos los cambios afectivos y cognitivos.
CIRCUITOS DE REGULACIÓN DE LA FUNCIÓN CORTICAL Y SU RELACIÓN CON LA DEPRESIÓN POSTISQUEMIA CEREBRAL
En los últimos años se han logrado avances notables en el conocimiento de los circuitos y la neuroquímica de los núcleos de la base y de otras estructuras de la región basal del telencéfalo, incluyendo sectores límbicos. Lo anterior ha permitido consolidar el concepto que los núcleos de la base, la corteza cerebral y el tálamo interactúan a través de un conjunto de circuitos paralelos encargados de procesar información no sólo motora, sino cognitiva y emocional.
Según Parent (87,'88) se reconocen circuitos cortico-estriado-tálamo-corticales de las siguientes categorías: motor, oculomotor, prefrontal dorsolateral, prefrontal medial, orbitofrontal y límbico estriatal. El circuito motor, el más conocido, comunica la corteza motora con el putamen, a este con el globo pálido externo e interno, a estos con los núcleos motores del tálamo ventral anterior y ventral lateral, que se proyectan a la corteza motora. El circuito oculomotor juega un papel importante en la estabilización y coordinación de los movimientos oculares; se origina en el área 8, que se proyecta al putamen, globo pálido, núcleos intralaminares del tálamo y de estos al área 8. El circuito prefrontal dorsolateral se asocia a las funciones mentales superiores, entre estas a la memoria operativa o "working memory"; se inicia en la corteza prefrontal lateral, se proyecta al núcleo caudado, de este a la sustancia negra reticulada localizada en el mesencéfalo y de allí al núcleo dorsomediano del tálamo que retroalimenta la corteza prefrontal. Los circuitos prefrontal medial y orbitofrontal relacionados con la regulación de la motivación y la función emocional respectivamente, se proyectan a regiones ventrales del estriado y del núcleo accumbens, de estos al pálido ventral, a la región límbica de la sustancia negra reticulada y de esta a las regiones mediales del núcleo dorsomediano que los proyecta a las cortezas de origen. El eje del sistema límbico conformado por el hipocampo y la amígdala se proyecta al núcleo accumbens, desde allí a las regiones límbicas de la sustancia negra reticulada, a los núcleos talámicos de la línea media, quienes, a su vez, devuelven a las áreas límbicas del lóbulo temporal. La función de uno o varios de estos circuitos puede estar comprometida en diferentes patologías como la enfermedad de Parkinson, Corea .de Hungtinton, esquizofrenia, trastorno obsesivo compulsivo y depresión.
Las proyecciones corticoestriatales procedentes de áreas límbicas y neocorticales son glutamatérgicas y por lo tanto excitatorias. Su activación promueve la excitación de células gabaérgicas localizadas en el estriado dorsal, el estriado ventral o el núcleo accumbens. Las anteriores provocan la inhibición de células gabaérgicas del globo pálido y de la sustancia negra cuya descarga es tónica. Dado que estas se proyectan al tálamo, su inhibición promovería un influjo excitatorio tálamocortical correspondiente a los sitios corticales en los cuales se inició la actividad. Se propone que la coincidencia o el acople entre las regiones tálamocorticales desinhibidas y las regiones corticales que iniciaron la actividad motora, cognitiva o emocional es esencial para el funcionamiento apropiado. Un desacople en el circuito facilita alteraciones del movimiento, de la actividad cognitiva o de la función emocional. Las proyecciones corticoestriatales y tálamocorticales, de carácter excitatorio, que utilizan el glutamato como neurotransmisor, interactúan con receptores NMDA, AMPA, KAINATO y metabotróficos. El neurotransmisor GABA caracteriza a los circuitos intraestriatales (estriato-palidales y estriatonigrales), pálido-talámicos, pálido-reticulares y nigro-talámicos procedentes de la sustancia negra reticulada; también participan interneuronas colinérgicas a lo largo del estriado. El equilibrio en la corteza cerebral entre las acciones excitatorias de las aferentes córtico-corticales y tálamo- corticales y las inhibitorias a través de las neuronas gabaérgicas (células en cesta, células en candelabro y células de doble bouquet), que en conjunto determinan la descarga de las células de proyección asociativas y comisurales de la corteza cerebral, está bajo la regulación modulatoria de varios sistemas de neuronas que se proyectan desde el tallo cerebral y desde regiones básales del telencéfalo.
Estos sistemas comprenden proyecciones dopaminérgicas (89-90), noradrenergicas (91), serotoninérgicas (91,92) y colinérgicas (92). Las primeras proceden de la porción dorsal de la sustancia negra y del área tegmental ventral (VTA) y en su conjunto constituyen el denominado sistema dopaminérgico mesolímbico y mesocortical. Las noradrenergicas proceden del locus coeruleus; las proyecciones serotoninérgicas se originan en los núcleos del rafe mesencefálico y las colinérgicas de los núcleos básales de Meynert. Por lo tanto, la salida neta de un conjunto de módulos corticales es el resultado de las complejas interacciones neuroquímicas entre las acciones excitatorias (glutamatérgicas), inhibitorias (gabaérgicas) y la regulación sobre ambas de los sistemas de proyección de origen subcortical. A su vez, las neuronas de origen de estos sistemas reciben proyecciones glutamatérgicas de la corteza (93,94).
Como hemos señalado, los núcleos de la base, esencialmente conformados por neuronas gabaérgicas de tipo espinoso mediano, reciben su mayor número de aferentes glutamatérgicas (excitatorias) de la corteza cerebral incluidas las áreas neocorticales límbicas y paralímbicas. La salida neta de los núcleos de la base hacia sus sitios de proyección es de tipo gabaérgica y por tanto inhibitoria. De manera análoga a lo que ocurre en la corteza, las neuronas del estriado, del pálido y de la sustancia negra reticulada, están inervadas por los sistemas modulatorios dopaminérgico, serotoninérgico, norepinefrínico y localmente por interacciones colinérgicas. Las dos primeras con una influencia mayor en cuanto a densidad y de gran importancia desde el punto de vista farmacológico.
Los sistemas de fibras modulatorios hacia la corteza cerebral y el estriado que proceden del tallo cerebral cursan por la porción ventral del encéfalo y abordan a los núcleos de la base, putamen y caudado por su superf ice ventral. Los que continúan hacia la corteza se dirigen hacia el polo frontal y se ramifican cefalocaudalmente para extenderse por toda la corteza, con un predominio de la inervación dopaminérgica en las regiones prefrontales y temporales, con mayor densidad en la corteza motora, la norepinefrínica en la corteza somatosensorial y la serotoninérgica en la corteza visual. La acumulación de estas fibras en las regiones ventrales del núcleo caudado y putamen en tránsito hacia la corteza frontal anterior puede estar en relación con la mayor frecuencia de alteraciones en la esfera emocional que se aprecia en pacientes con lesiones en las regiones anteriores del lóbulo frontal en comparación con lesiones de localización posterior. No está establecido si estos sistemas de proyección son asimétricos (distintos en los dos hemisferios), lo cual podría ser importante para explicar la aparición de depresión en lesiones del hemisferio izquierdo. Sin embargo, es importante anotar que de acuerdo con los estudios de Grasso y colaboradores (55) si una lesión isquémica involucra estructuras subcorticales en cualquiera de los dos hemisferios se puede presentar depresión como factor común. Aun así, lesiones en el hemisferio derecho pueden ir acompañadas de manifestaciones maníacas (11,13).
Teniendo en cuenta las apreciaciones neuroanatómicas y fisiológicas descritas, se puede postular que en las lesiones de la corteza cerebral por un evento isquémico y del estriado por efecto directo de la isquemia o denervación por la lesión cortical, se suspenden los impulsos gabaérgicos inhibitorios estriatonigrales ocasionando, con el transcurso de los días, un fenómeno de sobreregulación de los receptores GABA y de benzodiazepinas en la sustancia negra. Por otro lado, el efecto excitatorio por parte del núcleo subtalámico, no equilibrado por la función inhibitoria estriatal, podría generar un fenómeno de sobreexcitación con consecuencias metabólicas disfuncionales para las neuronas, las cuales se detectan por los cambios en la inmunoreactividad a la MAP2 en la sustancia negra.
Por su parte, la sustancia gris periacueductal, que como se planteó presenta cambios en la inmunoreactividad después de una lesión isquémica cortical, podría relacionarse con algunas de las manifestaciones clínicas afectivas si admitimos que un cambio similar podría ocurrir en el ser humano. La sustancia gris periacueductal tiene profusas conexiones con estructuras del tallo, del diencéfalo y del telencéfalo, especialmente límbicas. Fisiológicamente se ha relacionado con el control autonómico, conductas defensivas, aversión y miedo(95), situaciones éstas asociadas a las funciones del sistema límbico, tanto que se propone a la sustancia gris periacueductal como un subsistema de las estructuras límbicas (96).
Los cambios en la reactividad a la proteína MAP2 y en los receptores de benzodiazepinas de la sustancia negra y la alteración de la MAP2 en la sustancia gris periacueductal, podrían tener efectos sobre las expresiones emocionales posteriores a lesiones cerebrales corticales. Los fenómenos exofocales que se pueden interpretar como cambios en la conectividad, pueden perpetuarse por el desencadenamiento de procesos apoptóticos (97), fenómenos que podrían relacionarse con las manifestaciones clínicas crónicas que se suceden por una lesión vascular cerebral.
ASPECTOS FARMACOLÓGICOS DE LA DEPRESIÓN POSTISQUEMIA CEREBRAL
GENERALIDADES
Teniendo en cuenta que las manifestaciones depresivas posteriores a una isquemia cerebral presentan una historia natural diferente si se trata de una lesión izquierda o derecha, seguramente la aproximación terapéutica podría variar en uno u otro caso. Sin embargo, hasta el momento no existen estudios que evalúen diferencialmente la respuesta de los pacientes con lesiones cerebrales según el hemisferio afectado, así como tampoco hay informes de seguimiento con tratamiento antidepresivo a largo plazo. En general, son pocos los trabajos que han estudiado sistemáticamente el efecto de los antidepresivos en la depresión postisquemia cerebral y su comparación es difícil por heterogeneidad de las poblaciones evaluadas y al no considerar lo particular de las lesiones presentadas.
En varios estudios con nortriptilina (98,99) se ha observado una buena respuesta al tratamiento inicial, que desafortunadamente con frecuencia obliga a su suspensión por los efectos colaterales del fármaco. Con el uso de trazodone(100), también se ha comprobado una buena respuesta en la recuperación de las actividades cotidianas, pero igualmente los efectos colaterales fueron importantes especialmente por la edad avanzada del grupo estudiado.
Entre los antidepresivos inhibidores de la recaptura de serotonina se ha señalado la eficacia clínica del citalopram (101) y la fluoxetina (98,102) en la depresión postisquemia cerebral. Los efectos secundarios son menos comunes que con los antidepresivos tricíclicos, principalmente la ausencia de sedación y de efectos anticolinérgicos, antihistamínicos y cardiotóxicos. Sin embargo, se ha informado náuseas y vómito con frecuencia durante la primera semana de tratamiento.
LOS ANTIDEPRESIVOS Y EL SISTEMA DE RECEPTORES N-METIL-D-ASPARTATO (NMDA)
Aunque se tiene el concepto que el efecto clínico de los antidepresivos es el resultado de su acción sobre los sistemas de monoaminas y serotonina, en los últimos años se ha planteando la posibilidad de la mediación de los receptores de glutamato tipo N-metil-D-aspartato (NMDA). Investigaciones preclínicas realizadas por Layer y colaboradores (103) muestran como el tratamiento crónico con antidepresivos puede ocasionar cambios adaptativos en los receptores NMDA, cambios que sugieren un mecanismo de acción a través de este sistema de receptores y plantea la hipótesis del papel del glutamato en la fisiopatología de la depresión.
El efecto más conocido de los antidepresivos sobre el sistema de receptores NMDA está dado por la reducción que estos fármacos ocasionan sobre la afinidad establecida de sustancias como los antagonistas no competitivos dizocilpine y MK-801, el antagonista competitivo de alta afinidad CGP 39653 y el antagonista específico de alta afinidad del receptor del coagonista glicina, ácido 5,7 dicloroquinuréico (103,104) La disminución de la afinidad a estas sustancias podría corresponder a un cambio del receptor NMDA con repercusiones en la fisiología general de las neuronas piramidales de la corteza cerebral, pues se sabe que ocurre solamente allí y no en otras áreas del sistema nervioso como el hipocampo, el cuerpo estriado o regiones básales del telencéfalo. Se desconoce el mecanismo mediante el cual los antidepresivos modulan la acción de los receptores NMDA y se plantea que el uso crónico de estos fármacos puede inducir modificaciones en la composición o cambios estequiométricos en las subunidades que conforman el receptor NMDA.
Adquiere especial interés el hecho que la serotonina produzca un efecto en la liberación de glutamato (ligando natural de los receptores NMDA), un aminoácido que en concentraciones superiores a las fisiológicas (como ocurre en las fases aguda y subaguda de la isquemia cerebral), se comporta como neurotóxico al ocasionar cambios en la permeabilidad de las membranas a diferentes iones, especialmente al calcio(56). La activación de los subtipos de receptores serotoninérgicos SHTj y 5HT2 disminuye la liberación de glutamato en el espacio sináptico, lo que sugiere un papel protector de la serotonina sobre el fenómeno excito tóxico.
Aunque se conoce que la función básica del sistema serotoninérgico en el cerebro consiste en modular directa o indirectamente la actividad de las neuronas piramidales de la corteza cerebral y de las neuronas espinosas medianas en el cuerpo estriado(91), se observan respuestas variadas de los diferentes subgrupos de receptores a su agonista natural. Es así como se ha establecido que los receptores postsinápticos 5HT1A y 5HT2 ejercen un efecto inhibitorio y excitatorio respectivamente (53). Consecuentemente, se ha descrito el efecto neuroprotector de agonistas de receptores 5HT, (105,106) y de antagonistas de receptores 5HT2 (106,110) en animales de experimentación.
De otra parte el citalopram, una molécula inhibidora de la recaptura de serotonina, ha mostrado efectos neuroprotectores en circunstancias de isquemia cerebral experimental en ratas, ya que tiende a normalizar las concentraciones, usualmente incrementadas, de serotonina (102) y su metabolito ácido 5-hidroxiindolacético (5-HIAA) (111). El mecanismo preciso de su efecto neuroprotector es desconocido y se relaciona con su repercusión en el sistema serotoninérgico que, como hemos visto, implica cambios en receptores como el NMDA relacionado con fenómenos excitotóxicos y de muerte neuronal.
En conclusión, el efecto de los antidepresivos sobre la sensibilidad de los receptores NMDA a diferentes ligandos, la repercusión que ejercen en la excitotoxicidad ocasionada por la disminución de la liberación de glutamato, las consecuencias de la normalización de las concentraciones de metabolitos de serotonina y sobre los receptores postsinápticos 5HT1A y 5HT2 podrían traducirse en neuroprotección. En concordancia con estos planteamientos se ha demostrado que la fluoxetina, la maprotilina, el citalopram y la nortriptilina producen un resultado terapéutico no solo antidepresivo, sino también en la recuperación neurológica, la recuperación de las habilidades funcionales y la función cognoscitiva (92-95). Darnm y colaboradores (102) muestran que pacientes hemipléjicos sin depresión postisquemia cerebral medicados con fluoxetina y maprotilina presentan mejor respuesta a la terapia física si se comparan con pacientes en iguales condiciones que no toman alguno de estos medicamentos. Este estudio clínico postula que los antidepresivos tienen consecuencias farmacológicas que trascienden su conocido efecto antidepresivo.
CONCLUSIÓN
La alta prevalencia de la depresión postisquemia cerebral, su caracterización clínica, especialmente su evolución temporal diferencial relacionada con el hemisferio afectado, aspecto éste congruente con los estudios experimentales y funcionales cerebrales, permiten afianzar la idea de la depresión postisquemia cerebral como un trastorno depresivo orgánico.
Aunque una lesión isquémica cerebral no comprometa directamente las estructuras límbicas o de regulación emocional, los datos presentados por diferentes autores y por nuestro grupo, en los que se establecen cambios en receptores de benzodiazepinas, en la proteína asociada a microtúbulos (MAP2) y en la proteína acida glial fibrilar (GFAP) en sectores alejados del foco isquémico y relacionados con las estructuras límbicas como la sustancia negra o la sustancia gris periacueductal, permiten señalar una hipótesis fisiopatológica de la depresión postisquemia cerebral. En ella se encuentran implicados los circuitos córtico-estriato-tálamo-corticales y los diferentes sistemas de modulación provenientes del tallo cerebral como la serotonina, la norepinefrina, la dopamina y la acetilcolina.
Se empieza a considerar que la efectividad de los antidepresivos en la depresión postisquemia cerebral no está exclusivamente relacionada con el efecto de estos sobre la función aminérgica, sino que producen modificaciones en los receptores NMDA corticales y en los neurotransmisores como el glutamato, que en situaciones patológicas se encuentran implicados en procesos de excitotoxicidad. Queda por determinar el efecto neuroprotector de los antidepresivos, lo que hace necesario realizar más pruebas preclínicas y clínicas.
AGRADECIMIENTOS
Los autores expresan sus agradecimientos a la Vicedecanatura de Investigaciones de la Facultad de Salud, a la Vicerrectoría de Investigaciones por su apoyo para la ejecución de este trabajo y a los técnicos Martha Ceballos y Diego Pérez por su asistencia.
REFERENCIAS
1. Kasner E, Saver J, Levine S. Highiighs from the 25th Annual International Stroke Conference February 10-12 2000 [ Links ]
2. World Health Organization Task Forcé on Stroke preventions and other Cerebrovascular Disorders: recommendation on stroke preventions, diagnosis and therapy. Stroke 1989; 20:1407-1431 [ Links ]
3. Dam H, Pedersen HE, Ahlgren P. Depression among patients with stroke. Acta Psychiatr Scand 1989; 80: 118-24 [ Links ]
4. Fuh JL, Liu HC, Wang SJ, Llu CY, Wang PN. Poststroke depression among de chínese elderly ¡n a rural communily. Stroke 1997; 28:1126-1129 [ Links ]
5. Morris PL, Robinson RG, de Carvalho ML, Albert R Wells JC, Samuels JF, Eden-Fetzer D, Pnce TR. Lesión characteristics and depressed mood in the stroke data bank study. J Neurpsy and Clin Neurosci 1996; 8:153-159 [ Links ]
6. Morris PL, Robinson RG, Raphael B, Hopwood M.J. Lesión location and poststroke depression. J Neurpsy and Clin Neurosci 1996; 8:399-403 [ Links ]
7. Philip RR Morris MD, Robinson RG, de Carvalho ML, Albert R Wells J, Samuels J, Eden-fetzer D.; PriceT.R. Lesión, characteristics and depressed mood in the stroke data bank study. Journal of Neuropsychiatry and clinical neurosciences 1996; 8: 1 53-159 [ Links ]
8. Sorensen PS, Marquardsen J, Pedersen H, Helberg A, Munck O. Long-term prognosis and quality of life after reversible cerebral ischemic attack. Acta Neurol Scand 1989; 79: 204-213 [ Links ]
9. Astróm M. Generalized anxiety disorder in stroke patients : a 3-year longitudinal study. Stroke 1996; 27 :270-275 10. Fujikawa T, Yamawaki S, Touhouda Y. Silent cerebral infarction in patients with late-onset mania. Stroke 1995; 26 :946-949 [ Links ] [ Links ]
11. Berthier ML, Kulisevsky J, Gironell A, Fernández Benites JA. Poststroke bipolar affective disorders and anatomical correlates. J Neurpsy and Clin Neurosci 1996; 8 :160-167 [ Links ]
12. Robinson RG, Starkstein SE. Current research ¡n affective disorders following stroke. J. Neuropsychiatry Clin Neurosci 1990; 2:1-14 [ Links ]
13. Robinson R, Travella J. Neuropsychiatry of mood disorders. Neuropsychiatry, Ed Wiliams & Wilkins New York 1996. 287-305 [ Links ]
14. Parikh RM, Robinson RG, Lipsey JR. The impact of poststroke depression on recovery in activities of daily living over 2 year follow-up. Arch Neurol 1990; 47: 785-789 [ Links ]
15. Angeleri F, Angeleri VA, Foschi N, Giaquinto S, Nolfe G. The influence of depression, social activity, and family stress on functional outcome after stroke. Stroke 1993; 24:1478-1483 [ Links ]
16. Fujikawa T, Yanai Y, Yamawaki S. Psychosocial stressor in patients with major depression and silent cerebral infarction. Stroke 1997; 28: 1123-1125 [ Links ]
17. Astróm M, Adolfsson R, Asplund K. Major depression in stroke patients : 3-year longitudinal study. Stroke 1993; 24 :976-982 [ Links ]
18. Fujikawa T, Yamawaki S, Touhouda Y. Incidense of cerebral infarction ¡n patients with major depression 1993; 24 :1631-1634 [ Links ]
19. House A, Dennis M, Warlow C. Mood disorders after stroke and their relation to lesión location. Brain 1990; 113: 1113-1129 [ Links ]
20. Kahuanen M, Korpelainen J, Hiltunen R Brusin E, Mononen H, Maata R, Nieminen R Sotaniemi K, Myllyla V. Poststroke depressión correlates with cognitive impairment and neurological déficit. Stroke 1999; 30: 1875-1880 [ Links ]
21. Pohjasvaara T, Leppavuori A, Siira I, Vataja R, Kaste M, Erkinjuntti T. Frequency and clinical determinants of poststroke depresión. Stroke 1998; 11: 2311-2317 [ Links ]
22. Philip RR Morris MD, Robinson RG, Raphael B, Hopwood M.J. Lesión, location and poststroke depresión. The Journal of Neuropsychiatry and clinical neurosciences 1996; 8: 399-403 [ Links ]
23. Herrmann M, Bartels C, Schumacher M, Wallesch CW. Poststroke depression : ¡s there as pathoanatomic correlate for depression in the postacute stage of stroke ?. Stroke 1995; 26: 850-856 [ Links ]
24. House A, Dennis M, Mogridge L, Waelow C, Hawton K, Jones L. Mood disorders in the year after first stroke. Br J Psychiatry 1991; 158: 83-92 [ Links ]
25. Santus G, Ranzenigo A, Caregnato R, Inzoli MR. Social and family íntegration of hemispheric hemiplegic elderly patients 1 year after stroke. Stroke 1990; 21: 1019-1022 [ Links ]
26. Wade DT, Legh-smith J, Hewer RA. Depressed mood after stroke : a community study of ¡ts frequency. Br J Psychiatry 1987; 151: 200-205 [ Links ]
27. Murrel SA, Himmelfarb S, Wright K. Prevalence of depression and its correlates ¡n older adults. Am J Epidemiol 1983; 117: 173-185 [ Links ]
28. Sjogren K. Leisure after stroke. Int Rehabil Med 1982; 4: 80-87 [ Links ]
29. Robinson RG, PriceTR. Post-stroke depressive disorders : follow up study of 103 patients. Stroke 1982; 13: 635-641 [ Links ]
30. O.M.S. Clasificación Internacional de las Enfermedades (CIE10) 1996 [ Links ]
31. A.PA. (American Psychiatric association). Diagnostic and Statistical Manual of Mental Disorders Fourth Edition DSM 4. Washington DC, 1994 [ Links ]
32. Lipsey JR, Spencer WC. Rabins R Phenomenological comparison of poststroke depression and function depression. Am J Psychiatry 1986; 143: 527-529 [ Links ]
33. O'Rourke S, Lewis M, Sharpe C. Emotional outcomes after stroke: factors associated with poor outcome. J Neurol Neurosurg Psych 2000; 68: 47-52 [ Links ]
34. Choi-Kwon, Kim J. Sexual activities and emotional incontinence in stroke patients. Stroke 2000; 31: 302-305 [ Links ]
35. Robinson R. Neuropsychiatric consequences of stroke. Annu Rev Med 1997; 48:217-229 [ Links ]
36. Starkstein SE, Fedoroff R Price TR, Leiguarda R, Robinson R. Apathy following cerebrovascular lesions. Stroke 1993; 24: 1625-1630 [ Links ]
37. House A, Dennis M, Molineus. Emotinalism after stroke. Br Med J 1989; 298: 991-994 [ Links ]
38. Shimoda K, Robinson R. The relationship between poststroke depression and lesión location in long-term follow-up. Biol Psichiatry 1999; 45: 187-192 [ Links ]
39. Hermann N, Black S, Lawrence J. A prospective study of depressive symptoms and functionel outcome. Stroke 1998; 29: 614-624 [ Links ]
40. Andersen G, Vestergaard K, Ingemann-Nielsen M. Risk factors for post stroke depression. Acta Psych Sacand 1995; 92: 193-198 [ Links ]
41. Herrmann M, Bartles C, Wallesch CW. Depression in acute and chronic aphasia. J Neurol Neurosurg Psychiatry 1993; 56:672-678 [ Links ]
42. Mosnik DM, Williams LS, Welch R. The neuroanatomy of post-stroke depression and quality of life. Stroke 2000; 31: 299-301 [ Links ]
43. Lai S-M, Duncan PW, Keighley J. Depression and stroke recovery. Stroke 2000;31:292-298 [ Links ]
44. House A. Depression associated with stroke. J Neurol Neurosurg Psych 1996; 64: 14-22 [ Links ]
45. Gainotti G. Emotional behavior and hemispheric side of the brain. Cortex 1972; 8:41-45 [ Links ]
46. Robinson R, Trvella J. Neuropsychiatry of mood disorders. Neuropsychiatry, Editorial Wiiliams & Wilkins 1997; 287-305 [ Links ]
47. Sinyor D, Jacques R Kaloupek DG. Post stroke depression and brain location. Brain 1986; 109: 537-547 [ Links ]
48. Morris PL, Robinson RG, Raphael B. Lesión characteristics and post stroke depresión: evidence a specific relationship ¡n the left hemisphere. Neuropsychol and Behav Neurol 1992; 3:75-82 [ Links ]
49. Starkstein S, Robinson R, Berthier M, Price T. Depressive disorders following posterior circulations as compared to middle cerebral artery infarct. Brain 1988;11:375-387 [ Links ]
50. Phelps ME, Mazziota JC, Baxter L. Positrón emisión tomographic study of affective disorders: problems and strategies. Ann Neurol 1984; 15 (suppl): 149-156 [ Links ]
51. Inchausti L, González MA. Imágenes cerebrales en la depresión. Neuropsiquiatría, Imágenes del Cerebro y la Conducta Humana, Moisés Gaviria y Jorge E Téllez. Editorial Nuevo Milenio 1995; 287-308 [ Links ]
52. Mayberg HS, Robinson RG, Wong DR. PET imaging of cortical S2 serotonin receptors after stroke: lateralized changes and relationship to depression. Am J Psychiatry 1988; 145: 937-943 [ Links ]
53. Leysen JE, Commeren W, Schote A. Serotonin receptor subtypes: possible roles and ¡mplications ¡n antipsichotic drug action. Serotonin ¡n antipsichotic treatment. Editorial Dekker 1996; 77-107 [ Links ]
54. Starkstein S, Robinson R, Berthier M. Differential mood changes following basal ganglia vs thalamic lesión. Arch Neurol 1988; 45: 725-730 [ Links ]
55. Grasso MG, Pantano R Ricci M, Intiso DF, Pace A, Padovani A, Orzi F, Pozilli C, Lenzi GL. Mesial temporal cortex hipoperfusion in associated with depression ¡n subcortical stroke. Stroke 1994; 25: 980-985 [ Links ]
56. Lee JM, Zipfel G, Choi D. The changing landscape of ¡schaemic brain injury mechanism. Nature 1999; 399: A7-A9 [ Links ]
57. Schwartz RD, Yu X, Katzman MR, Hayden-Hixson DM, Perry JM. Diazepam, given postischemia, protects selectively vulnerable neurons in the rat hippocampus and striatum. J Neurosci 1995; 15:529-539 [ Links ]
58. Sternau L, Lust WT, Ricci AJ, Ratcheson R. Role for gama-aminobutync acid in selecive vulnerability in gerbils. Stroke 1989;20:281-287 [ Links ]
59. Sugimoto A, Takeda A, Kogure K, Onodera H. NMDA receptor (NMDAR1) expression in the rat hippocampus after forebrain ischemia. Neurosci Let 1994; 170: 39-42 [ Links ]
60. Ginsberg MD, Bustos R. Rodent models of cerebral ischemia. Stroke 1989; 20: 1627-1642 [ Links ]
61. Hallmayer J, Hossmann KA, Mies G. Low doses of barbiturates for preventions of hippocampal lesions after brief ischemic episodes. Acta Neuropathol 1985; 68: 27-31 [ Links ]
62. Kimelberg H, Noremberg M. Astrocytes. Books of Scientific American, Editorial Prensa Científica 1991; 18-27 [ Links ]
63. Vermuganti L, Raphavendra R, Adibhata M, Dofgan A, Bowen K, Hatcher J, Rothslein S, Dempsy R. Glial gluatmate transporter GLT-1 down-regulation precedes delayed neuronal deatn in gerbil hippocampus following transiant global cerebral ischemia. Neurochemistry International 2000; 36: 531-537 [ Links ]
64. Gegelashuili G, Schousboe R. High affinity glutamate transporters: regulation of expression and activity. Mol Pharm 1997; 52: 6-15 [ Links ]
65. Andrew R. Transhemispheric diaschisis. A review and comment. Stroke 1991; 22: 943-949 [ Links ]
66. Belayeb L, Zhao W, Busto R, Ginsberg M. Transient middle cerebral artery occlusion by ¡ntraluminal suture: I. Three-diernensional autoradiographic image-analysis of local cerebral glucose metabolism-blood flow interrelationships during ischemia and early recirculation. J cereb Blood Flow Metab1997; 12: 1267-1280 [ Links ]
67. Shao W, Belayeb L, Ginsberg M. Transient middle cerebral artery occlusion by ¡ntraluminal suture: II. Neurological déficit, and pixel-based correlation of histopathology with local blood flow and glucose utilization. J cereb Blood Flow Metab 1997; 12: 1281-1290 [ Links ]
68. Belayeb L, Alonso OF, Busto R, Zhao W, Ginsberg MD. J Cereb Blood Flow and Metab occlusion ¡n the rat by ¡ntraluminal suture. Stroke 1996; 27: 1616-1623 [ Links ]
69. Longa EZ, Wenstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20: 84-91 [ Links ]
70. Arango C, Escobar M, Rocha L, Pimienta H. Alteración de los sistemas de neuronas excitatorias e inhibitorias en la corteza cerebral de ratas evaluados en un modelo de isquemia cerebral. Simposium International Restauraron Neurológica Ciudad de la habana, febrero de 1999 [ Links ]
71. Pimienta H, Arango C, Pedroza A, Escobar M. Respuesta neurobiológica a la lesión cerebral isquémca. Foco,penumbra y regiones exofocales Neurociencias en Colombia. En prensa [ Links ]
72. Arango C, Rocha L, Escobar M, Pimienta H. Cambios exofocales en la expresión de receptores de benzodiazepinas y de la protelna asociada a microtúbulos en la sustancia negra inducido por oclusión reversible de la arteria cerebral media en ratas. Simposium International Restauration Neurológica Ciudad de la Habana, Cuba. Febrero de 1999. pp 119. [ Links ]
73. Unnerstall JR, Kuhar MJ, Niehoff DL, Palacios JM. Benzodiazepine receptors are coupled to a subpopulation of gamma-aminobutyric acid (GABA) receptors : evidence from a quantitative autoradiographic study. J Pharmacol Exp Ther 1981; 218: 297-804 [ Links ]
74. Enna SJ, Móhler H. gamma-aminobutyric acid (GABA) receptors and their association with benzodiazepine recognition sites. Psychopharmacology : The Third Generation of Progress. Edited by Herbert Y. Meltzer. New York: Raven Press; 1987: 265-272 [ Links ]
75. Kanai Y, Hirokawa N. Sorting mechanism of Tau and MAP2 in neurons : suppressed axonal transot of MAP2 and locally regulated microtubule binding. Neuron 1995; 14: 421 -432 [ Links ]
76. Kitagawa K, Matsumoto M, Nünobe M, Micoshiba K, Hata R, Ueda H, Manda N, Fukunaga R, Isaka Y, Kimura K, Kodama T. Microtubule-associated protein 2 a sensitive marker for cerebral ¡schemic damage, ¡mmunohistochemical ¡nvestigation of dendritic damage. Neuroscience 1989; 31:401-411 [ Links ]
77. Miyazawa T. ; Bonnekoh R ; Hossmann K.A. Temperatura effect of immunostaining of Microtubule-associated protein 2 and synaptophysin after 30 minutes of forebrain ¡schemia in rat. Acta Neuropathol 1991 ;85 :526-532 [ Links ]
78. Yanahiara T, Brengman JM, Mushynki WE. Differential vulnerability of microtubule components in cerebral ischemia. Acta Neuropathol 1990; 80: 499-505 [ Links ]
79. Postmantur RM, Kampel BA, Liu SJ, Heck K, Taft WC, Clifton GL, Mayes RL. Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats : an immunofluorescence study. J Neuropathol and exp neurol 1996; 55: 68-80 [ Links ]
80. Ayala J, Escobar M. Efecto sobre la proteina asociada a microtúbulos MAP2 en la corteza frontal de rata inducidos por lesión aguda con acido kaínico intracortíocal. Simposio Restauración Neurológica. 1999. La Habana, Cuba. pp119. [ Links ]
81. Nagasawa H, Kogure K. Correlation betwen cerebral blood flow and histologic changes in a new rat model of middle cerebral artery occlusion. Stroke 1989; 20: 1037-1043 [ Links ]
82. Nagasawa H, Araki T, Kogure K. Autoradiographic analysis of second-messenger and neurotransmitter receptor system in the exo-focal remote áreas of postischemic rat brain. Brain Res Bul 1994; 35: 347-352 [ Links ]
83. Nagasawa H, Araki T, Kogure K. Autoradiographic analysis of second messenger and neurotransmitter receptor bindings in the strionigral system of the postischemic rat brain. J Neurosci Res 1992; 33: 485-492 [ Links ]
84. Nagasawa H, Araki T, Kogure K. Alteration of muscarinic acetylcholine binding sites in the postischemic brain áreas of the rat using in vitro autoradiography. J Neurol Sci 1994; 121: 27-31 [ Links ]
85. Nagasawa H, Araki T, Kogure K. Alteration of dopamine D, receptor in the strionigrat system of the postischemic rat brain. Neurosci Let 1992; 134: 271-274 [ Links ]
86. Nagasawa H, Araki T, Kogure K. Alteration of adenosine A, receptor binding ¡n the post-ischaemic rat brain. Neuroreport 1994; 5: 1453-1456 [ Links ]
87. Parent A, Hazrati LN. Functional anatomy of the basal ganglia. I. The cortical-basal ganglia-thalamo-cortical loop. Brain Res Revi 1995; 20: 91-127 [ Links ]
88. Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Rev 1995; 20: 128-154 [ Links ]
89. Weiner I, Joel D. The connection of the dopaminergic system with the striatum in rats and primates: an analysis whit respect to the functional and comportamental organization of the striatum. Neurosc 2000; 96: 451 -474 [ Links ]
90. Goldman-Rakik PS, Muly EC, Williams G. D1 receptors in prefrontal cells and circuits. Brain Res Rew 2000; 31: 295-301 [ Links ]
91. Arango C, Ángulo E, Escobar MI, Pimienta JH. Sistema serotoninérgico y de la norepinefrina. Sistema Nervioso : Neuroanatomía Funcional, Neurohistología, Neurotransmisores, Receptores y Clínica. Editorial Universidad del Valle, Segunda Edición. 1998:245-253 [ Links ]
92. Wilson CJ. Basal Ganglia. Thesynaptic organization of the brain. 4a Edition Gordon M. Shepherd, Yale University School of Medicine U.S.A 1998; 229-275 [ Links ]
93. Bebió T, Wallesch C, Hermann M. The crucial role of frontostriatal circuits for depressive disorders in the postacute stage after stroke. Neuropsy Neuropsychol Behav Neurol 1999; 12: 236-246 [ Links ]
94. Escobar MI, Pimienta JH. Núcleos de la base. Sistema Nervioso : Neuroanatomía Funcional, Neurohistología, Neurotransmisores, Receptores y Clínica. Editorial Universidad del Valle, Segunda Edición. 1998 :119-136 [ Links ]
95. Bandler R, Shipley M. Columnar organization in the midbrain periaqueductal gray. modules for emotional expression. TINS 1994; 17: 379-389 [ Links ]
96. Escobar MI, Pimienta JH. Estructura interna del mesencéfalo. Sistema Nervioso : Neuroanatomía Funcional, Neurohistología, Neurotransmisores, Receptores y Clínica. Editorial Universidad del Valle, Segunda Edición. 1998 :197-206 [ Links ]
97. Linnik M, Zobrist R, Hatfield M. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 1993; 24: 2002-2009 [ Links ]
98. Gonzalez-Torrecillas JL, Mendlewics J, Lobo A. Effects of early treatment of poststroke depression on neuropsychological rehabilitation. Int Psychogeriatr 1995; 7: 547-560 [ Links ]
99. Lipsey JR, Pearlson GD, Robinson RG. Nortriptyline treatment of poststroke depression:a double blind study. Lancet 1984; 1:297-301 [ Links ]
100. Reding MJ, Orto LA, Winter SW. Antridepressant Therapy after stroke: a double blind trial. Arch Neurol 1986; 43: 763-765 [ Links ]
101. Andersen G, Vestergaard K, Lauritzen L. Effective treatment of poststroke depression with selective serotonin reuptake inhibitor citalopram. Stroke 1994; 25: 1099-1104 [ Links ]
102. Damm M, Tonin R De Boni A, Pizzolato G, Casson S, Ermani N, Freo U, Pirón L, Battistin L. Effects of fluoxetine on functional recovery in poststroke hemiplegic patients undergoing rehabilitation therapy. Stroke 1996; 27: 1211-1214 [ Links ]
103. Layer RT, Popik R Nowak G, Paul IA, Trullas R, Skolnik R A unified theory of antidepressant action: evidence for adaptation of the NMDA receptor following chronic antidepressant treatment. Ion chañéis pharmacology, edit Soria and Ceña, Oxford University Press 1998: 438-456 [ Links ]
104. Araki T, Kato H, Fujikawa T, Kogure K, Itoyama Y. Post-ischemic changes of [3H]glycine binding in the gerbil brain after cerebral ¡schemia. Eur J Pharmacology 1995; 278: 91-96 [ Links ]
105. Prehn JH, Backhaus C, Karcoutly C, Nuglish J, Peruche B, Rossberg C, Krieglstein J. Neuroprotective properties of 5HT1A receptor agonists in rodent models of focal and global cerebral ¡schemia. Eur J Pharmachol 199; 203: 213-222 [ Links ]
106. Prehn JH, Welsch M, Backhaus C, Karcoutly C, Nuglish J, Ausmeier F, Karcoutly C, Krieglstein J. Effects of serotoninergic drugs in experimental brain ¡schemia: evidence for a protective role of serotonin in cerebral ischemia. Brain Res 1993; 630: 10-20 [ Links ]
107. Fujikawa H, Kato H, Nakano S, Kogure K. A serotonin S2 antagonist, naftidrofuryl, exhibited a protective effect on ¡scherrric neuronal damage ¡n the gerbils. Brain Res 1989; 494:387-390 [ Links ]
108. Globus M, Wester R Busto R, Dietrich W. Ischemia induced extracellular reléase of serotonin. Stroke 1992; 23: 1595-1601 [ Links ]
109. Klisch J, Bode-Greuel K. Ketanserin reduces neuronal calcium accumulation and cell death in the hippocampus of the Mongolian gerbil after transient forebrain ischemia. Brain Res 1992; 578:1-7 [ Links ]
110. Takagi K, Gynsberg M, Globus M, Busto R, Dietrich D. The effect of ritanserin a 5HT2 receptor antagonist, on ischemic cerebral blood flow and infarct volume in rat middle cerebral artery occlusion. Stroke 1994; 25: 481 -486 [ Links ]
111. Nagao T, Ibayashi S, Sadoshima R Izumi J, Fujishima M. Citalopram, a serotonin reuptake inhibidor, and brain ¡schemia in SHR. Brain Res Bull 1995; 38: 49-52 [ Links ]

