










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.31 no.3 Bogotá July/Sept. 2002
Sistema glutamatérgico, primera parte: Sinaptología, homeostasis y muerte celular
Adriana M. Medina Marín, M. D. 1 Martha I. Escobar Betancourth, M. Se. 2
(1) Estudiante de Doctorado en Neurociencias, Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
(2) Profesora titular, investigadora del Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
Resumen
El ácido glutámico es el neurotransmisor más abundante del sistema nervioso. Las neuronas glutamatérgicas extienden su acción a lo largo del eje encefalomedular. Dos tercios de las neuronas de la corteza cerebral son glutamatérgicas. Las terminales glutamatérgicas establecen contactos en alta proporción con espinas dendríticas, estructuras sobre las cuales existen mayores indicios de plasticidad morfofuncional y que se asocian a los procesos integrativos más complejos. El papel del ácido glutámico y su disfunción ha ganado importancia en neurología y en psiquiatría, en la medida en que se ha profundizado en el conocimiento sobre su metabolismo, tipos de receptores, transportadores y mecanismos de homeostasis, cuya disfunción puede llevar a muerte neuronal.
Palabras claverácido glutámico, nmetilaspartato, ácido kaínico, sistemas de transporte de aminoácidos, apoptosis, síndrome de neurotoxicidad.
Abstract
The glutamic acid is the most abundant neurotransmitter oí the nervous system. The glutamatergic neurons spread its excitatory actions alog the CNS. Two thirds ofthe cortical neurons use glutamic acid as their transmitter. The glutamatergic endings primarily stablish synaptic contaets with dendritic spines, these structures play a major role in complex integrative functions and their activity is related to plasticity The synaptic glutamatergic function had deserved attention, considering its implication in Neurology and Psychiatry. In thepast fewyears a wealth of information has been accumulated in aspects ofthe glutamatergic system such as metabolism, types of receptors, transporters and homeostasis. Dysfunction ofthe glutamtergic system has been associated with a varíety of disorders that in some cases can be related to cell death.
Key Words: Apoptosis, glutamic acid, nmethyaspartate, kainic acid, aminoacid transport system, neurotoxicity syndromes.
Introducción
Las últimas dos décadas han per mitido un notable avance en el estudio del sistema de neuronas glutamatérgicas (figuras 1, 2 y 3); sin embargo, los principales hallazgos se refieren a la identificación de sus circuitos, organización sináptica, receptores, metabolismo, homeostasis y plasticidad. El presente artículo suministra una visión actualizada del sistema glutamatérgico, que pretende establecer las bases celular, subcelular y molecular, para aproximarnos a su comprensión y posible función en enfermedades neurológicas de curso agudo o crónico.
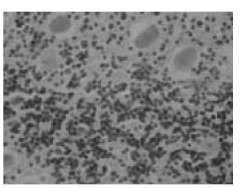
Figura 1. Corte de cerebelo La microfotografía de la Figura 1 corresponde a un corte de cerebelo, en el cual se observa, en la parte superior, la capa de células de Purkinje y, en la parte inferior de la imagen, la capa granular conformada por las células excitatorias del cerebelo (40X).
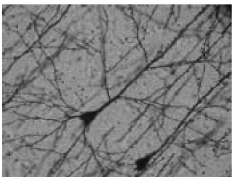
Figura 2. Corte de corteza cerebral La microfotografía de la Figura 2 corresponde a un corte de corteza cerebral, teñido con la técnica de Golgi; en ella se aprecian dos neuronas piramidales con sus árboles dendríticos básales y apicales, en los que se observan espinas. Los axones de ubicación inferior presentan colaterales recurrentes importantes en la integración de módulos intracorticales (40X).
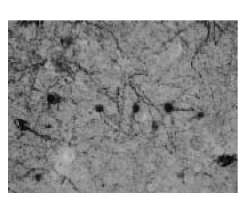
Figura 3. Corte del tálamo La microfotografía de la Figura 3 corresponde a un corte del tálamo, que muestra células glutamatérgicas positivas para parvalbumina (40X).
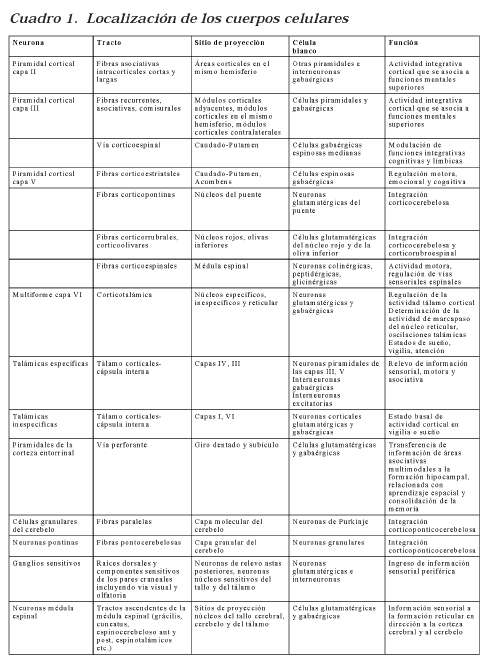
En el Cuadro 1 se muestra la localización de los cuerpos celulares, los tractos a que dan lugar, los sitios de terminación y las funciones que se sugieren para este tipo de neuronas.
Observe cómo las neuronas glutamatérgicas y sus fibras se extienden por amplios sectores del eje encéfalomedular y en el sistema nervioso periférico, para constituir vías ascendentes, descendentes, asociativas y microcircuitos locales.
Metabolismo del glutamato en el SNC
El glutamato cumple la mayoría de los criterios para ser considerado como neurotransmisor:
- Se localiza en vesículas presinápticas
- Se ha demostrado su liberación cuando se estimulan las terminales axónicas.
- Se han caracterizado receptores específicos que responden al ácido glutámico tanto en la membrana pre como en la postsináptica.
- Existen mecanismos para removerlo de la hendidura sináptica.
A pesar de lo anterior, en un principio fue algo difícil admitir el papel del glutamato como neurotransmisor, dado que esta molécula se encuentra ampliamente distribuida en las neuronas y en la glía y, además, se involucra en actividades consideradas no sinápticas. La principal de éstas es su participación en la función metabólica de las células. Por tal razón, es importante distinguir entre el pool de glutamato dedicado a la neurotransmisión y aquel que forma parte de las rutas metabólicas comunes a todas las células. Es necesario tener en cuenta que en las células gliales se convierte en glutamina, mientras que en las células gabaérgicas (en el caso de la corteza cerebral están en estrecha relación con las células glutamatérgicas piramidales), por acción de la enzima que decarboxila al ácido glutámico GAD, se convierte en el neurotransmisor Gaba (1).(2).
La glucosa sérica es el precursor más importante para la síntesis de ácido glutámico en el sistema nervioso; pues ingresa a éste luego de atravesar el endotelio vascular y la interfase (constituida por los pies de los astrocitos que rodean los capilares sanguíneos). Por lo tanto, la captura de glucosa se realiza por la acción de los transportadores de la familia GLUT, los cuales se expresan tanto en las células endoteliales como en los astrositos. Además, se ha comprobado que debido a la acción de estos astrocitos la glucosa se convierte en lactato, el cual se libera en el líquido extracelular, luego las neuronas lo captan y se constituye así en una de sus alternativas de sustrato energético.
Los astrocitos también poseen la astrocitos o en las neuronas, la glucapacidad de capturar ácido láctico tamina puede ser transferida de las de origen sérico, lo que probable glías a las neuronas; pues, por parmente los convierte en un importan te de los primeros, se han identifite reservorio de este metabolito. En cado dos transportadores que meconcentraciones elevadas, el ácido dian su liberación y tres transporláctico tiene un efecto ansiogénico tadores, localizados en las neuro(3),(4), lo que indica que en la ma ñas, que ayudan a atrapar esta susyoría de las circunstancias este efec tancia. to es normalmente amortiguado (sin embargo, además, tiene un efecto vasodilatador que puede ser un factor importante para correlacionar la expresan la enzima glutaminasa, la actividad glutamatérgica y el incremento del flujo sanguíneo cerebral).
Al contrario de las neuronas, los la membrana mitocondrial interna astrocitos tienen una capacidad li pero, en caso de lesión de las neumitada para sintetizar ácido lácti roñas, ésta puede trasladarse al co. Las neuronas, por otra parte, medio extracelular y, de manera convierten el lactato en piruvato y a aberrante, convertir la glutamina en éste en acetilCoA, el cual ingresa al glutamato en este sitio, lo cual puédelo del ácido tricarboxílico, donde de ser deletéreo para la célula por se une con el oxaloacetato para for los conocidos efectos excitotóxicos mar citrato. Éste pasa luego, sucesivamente, a isocitrato y a alfaceto glutarato, el cual puede ser transaminado a glutamato por transaminasas como la aspartato aminotransferasa y la alanina aminotransferasa, que utiliza donadores de grupos amino como aspartato y alanina (5).
En los astrocitos que rodean los cuerpos de las neuronas, es decir, los de localización en la sustancia gris y en los oligodendrocitos, el glutamato se convierte en glutamina por acción de la enzima glutamina sintetasa. Con la intervención de transportadores localizados en los astrocitos o en las neuronas, la glutamina puede ser transferida de las glías a las neuronas; pues, por parte de los primeros, se han identificado dos transportadores que median su liberación y tres transportadores, localizados en las neuronas, que ayudan a atrapar esta sustancia.
Se ha descrito que una subpoblación de neuronas glutamatérgicas expresan la enzima glutaminasa, la cual convierte la glutamina en lutamato. En condiciones normales la enzima glutaminasa se localiza en la membrana mitocondrial interna pero, en caso de lesión de las neuronas, ésta puede trasladarse al medio extracelular y, de manera aberrante, convertir la glutamina en glutamato en este sitio, lo cual puede ser deletéreo para la célula por los conocidos efectos excitotóxicos del glutamato en concentraciones ligeramente superiores a las normales. Esta puede ser una ruta importante para exacerbar y propagar un proceso de lesión excitotóxico (3).
Bajo ciertas condiciones, el transporte continuo de glutamina por el astrocito hacia la neurona conlleva una disminución de las concentraciones de alfacetoglutarato del ciclo del ácido tricarboxílico glial, lo cual se puede compensar utilizando la vía de la anaplerosis, es decir, la carboxilación del piruvato, para convertirlo en oxaloacetato o malato, e ingresar de ese modo nuevos sustratos al ciclo.
Por años se consideró que los astrocitos eran las únicas células capaces de llevar a cabo este tipo de metabolismo. Sin embargo, se ha demostrado la actividad carboxiladora del piruvato de la enzima málica mitocondrial neuronal. La posibilidad de que ciertas subpoblaciones neuronales puedan carboxilar el piruvato explica por qué algunos circuitos glutamatérgicos pueden expresar bajas concentraciones inmunoquímicas de glutaminasa. En este caso la producción de glutamato para la función sináptica se deriva de sustratos propios de estas neuronas (5).
Una vez el glutamato ha sido liberado en la sinapsis hay que removerlo de la hendidura para limitar su acción en el tiempo. Existen dos alternativas para manipular este glutamato: primero, que sea capturado por los astrocitos. Segundo, que sea capturado por las terminales
nerviosas y en las mitocondrias, organelas que abundan en estas estructuras, transaminado por aminotransferasas o deaminado por la
glutamato deshidrogenasa y convertido entonces a alfacetoglutarato, que es oxidado sucesivamente a succinato, fumarato y malato. Este
último puede ser descarboxilado a lactato, de manera que el glutamato de la neurotransmisión puede ser fuente de lactato. Esto es muy importante, porque este metabolito puede actuar en los vasos sanguíneos adyacentes como vasodilatador, y facilitar por esta vía un acople entre actividad neuronal y flujo sanguíneo local.
El ciclo de síntesis y degradación del glutamato requiere energía en varias de sus etapas: en el proceso de almacenamiento en vesículas y durante la fusión de la vesícula con la membrana presináptica, proceso previo para la exocitosis. Por otro parte, para la recaptura del glutamato y de la glutamina también se requiere energía, ya que este proceso debe vencer un gradiente de sodio que necesita la función de la Na/ K atpasa, que retorna el sodio al espacio extracelular. En esta actividad se necesitan dos ATP por cada molécula de glutamato o glutamina. Se calcula que el ciclo del glutamato consume alrededor del 3% del total de la energía obtenida del metabolismo de la glucosa (4).
Sinapsis glutamatérgicas de la corteza cerebral
Elementos presinápticos
El glutamato que se utiliza como glutamato deshidrogenasa y conver neurotransmisor se almacena en tido entonces a alfacetoglutarato, vesículas. Éstas lo atrapan con la que es oxidado sucesivamente a participación de moléculas que se succinato, fumarato y malato. Este localizan en la membrana vesicular, último puede ser descarboxilado a las cuales se conocen como translactato, de manera que el glutama portadores de glutamato vesicular to de la neurotransmisión puede ser (VGLUT). Así, el transportador infríente de lactato. Esto es muy im terioriza el glutamato con ayuda de portante, porque este metabolito un mecanismo que requiere interpuede actuar en los vasos sanguí cambio de protones; mientras que neos adyacentes como vasodilata la asociación entre las vesículas y el citoesqueleto permite su transporte hacia la membrana presináptica, de manera que se encuentren disponibles para la exocitosis (Figura 4).
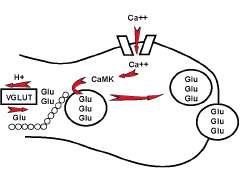
Figura 4. Transporte de glutamato hacia la membrana presináptica En la terminal presináptica el glutamato se acumula en vesículas, por medio del transportador VGLUT, el cual lo intercambia por protones. Las vesículas, inicialmente ancladas en el citoesqueleto, son liberadas por la actividad de la CaM kinasa, en respuesta al ingreso de calcio, y movilizadas hacia la terminal presináptica para liberar su contenido a la hendidura sináptica.
El proceso de liberación del glutamato es similar al de otros neurotransmisores. La liberación ocurre cuando la vesícula se fusiona con la membrana presináptica, por medio de la interacción con proteínas presentes en la membrana de las vesículas, en el citoplasma y en la membrana presináptica. Las proteínas mencionadas tienen la capacidad de formar un sistema de anclaje, que se activa con la entrada de calcio a la terminal presináptica. Hasta ahora sólo se han identificado algunas de las proteínas que participan en este proceso. En la membrana de la vesícula se reconocen entre otras, la sinaptobrevina, la sinaptofisina y la sinaptotagmina. En el citoplasma están presentes la alfaSNAP y la rab3, mientras que en la membrana presináptica se localizan la SNAP25 (proteína asociada a la sinápsis) y la fisofilina (6),(7),(8),(9),(1).
Las vesículas que se ligan al citoesqueleto por medio de la proteína sinapsina se liberan por la activación de la enzima calciocalmodulinakinasa (CAMkinasa), en respuesta al ingreso de calcio a la terminal presináptica, a su vez inducido por la llegada de un potencial de acción. Los iones de calcio facilitan la activación y organización de las proteínas de anclaje vesicular, del citoplasma y de la membrana presináptica. La interacción de estas proteínas forma un núcleo o eje que permite la fusión de la vesícula con la membrana presináptica, lo cual antecede a la exocitosis (Figura 5). Como la proteína rab está bajo la regulación de factores de crecimiento, este tipo de señales derivados de las glías o de otras neuronas pueden ser importantes para la regulación de los mecanismos de liberación del neurotransmisor y de la plasticidad sináptica asociada a los mecanismos de liberación (11).
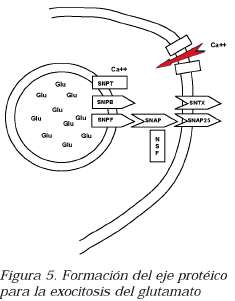
En la membrana vesicular, la sinaptotagmina (SNPT) actúa como sensor de Ca++ en la vesícula, la sinaptobrevina (SNPB) se encarga de reconocer la membrana presináptica y la sinaptofisina (SNPF) establece un poro de fusión con la membrana. En el citoplasma se encuentra la proteína de fusión NSF, que requiere la SNAP para su acople al eje. En la membrana presináptica se encuentra la sintaxina (SNTX), encargada de reconocer la membrana vesicular, y la SNAP 25, captadora de las proteínas citoplasmáticas.
Receptores del ácido glutámico
Una vez la vesícula se fusiona a la membrana presináptica, el neurotransmisor se libera en el espacio sináptico e interactúa con los receptores (ubicados tanto en la membrana pre como en postsináptica), que regulan esta operación. Téngase en cuenta que los receptores que res
ponden al ácido glutámico son de dos tipos: ionotróficos y metabotróficos. Los primeros incluyen en su estructura un canal por donde fluyen iones como calcio, sodio y potasio. Los metabotróficos no forman canales, pero están asociados al sistema de proteínas G y su activación promueve o inhibe el desencadenamiento de cascadas metabólicas que influyen en diferentes niveles, en la membrana, en el citoplasma e incluso en el núcleo de las neuronas. Es claro que las acciones ionotróficas desencadenan fenómenos eléctricos de instauración y activación rápida, mientras que las acciones metabotróficas son prolongadas y se pueden traducir en adaptaciones moleculares y cambios en la expresión génica, lo cual se considera importante en los mecanismos moleculares que subyacen a la plasticidad y a la memoria (12).
Es necesario aclarar que en ciertas circunstancias, por ejemplo, LTP (potenciación de largo plazo), sobreestimulación glutamatérgica, etc., la señalización mediada por el calcio a través de un canal ionotrófico puede llevar a la activación de cascadas de fosforilación, que pueden generar cambios perdurables en las neuronas receptoras.
Receptores ionotróficos del ácido glutámico
Los receptores ionotróficos son canales de cationes que cuando se abren, permiten la entrada de sodio y de calcio y la salida de potasio. Hay tres tipos y su nombre se deriva de sus agonistas sintéticos NMDA (NmetilDaspartato), AMPA (alfaamino3hidroxi5metil4isoxazole propionato) y kainato (véase Figura 6).
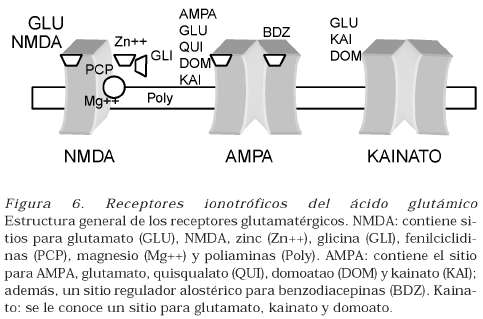
- Receptores NMDA: este receptor es estimulado por glutamato, NMDA y, en menor grado, por quisqualato y aspartato. Es permeable al calcio, al sodio y al potasio, por lo que la conductancia al primer ion es mayor. Los receptores tipo NMDA son grandes complejos proteicos que contienen, por lo menos, una de las ocho isoformas NR1 (AH), como subunidad fundamental, las cuales se pueden unir homoméricamente o de forma heteromérica con cualquiera de los subtipos de la subunidad NR2A a NR2D, para forman complejos de 45 subunidades (13), (14). La subunidad NR1 es un pequeño polipéptido que define las propiedades de este canal, el cual presenta bloqueo interno por magnesio dependiente de voltaje, bloqueo interno por zinc no dependiente de voltaje, un sitio externo de activación para poliaminas y requiere glicina como coagonista para su activación. Presenta un sitio de unión dentro del canal para sustancias modulatorias como los fármacos dizocilpine, fencyclidine, memantine y el anestésico ketamina. Además, cuenta con una región externa sensible a iones hidrógeno y un sitio redox constituido por uno o más grupos sulfidrilo, el cual puede ser oxidado por sustancias como el óxido nítrico. Este canal iónico no presenta desensibilización, lo cual lo hace electrofisiológicamente diferente de los demás receptores ionotróficos.
El receptor NMDA puede estar localizado tanto en la terminal pre como postsináptica, aun cuando es más abundante en esta última. Su papel presináptico no está muy claro, sin embargo, parece que regula la exocitosis del glutamato por medio de la entrada de calcio, incrementando y potenciando así la respuesta (13), (15).
- Receptores AMPA: este tipo de receptores posee un canal que media la más rápida aparición de un potencial postsináptico excitatorio después de la liberación de glutamato. Las subunidades que lo conforman están codificadas por una familia de cuatro genes, GLUR1 a GLUR4, que forman receptores heterologoméricos dentro de la membrana. Responden químicamente a quisqualato, AMPA, kainato y glutamato, pero no a NMDA y presentan un sitio modulatorio externo para cierto tipo de benzodiacepinas.
Dependiendo de la composición molecular de las subunidades de este tipo de receptor, el canal iónico asociado puede ser permeable al sodio, al potasio y posiblemente al calcio. Los receptores que carecen de la subunidad GLUR2 tienen un alto grado de permeabilidad al calcio. Se ha demostrado que en condiciones de isquemia, en la zona depenumbra hay una disminución en la expresión de la subunidad GLUR2. Esto conlleva a un incremento en la permeabilidad al calcio, lo cual podría amplificar el fenómeno excitotóxico mediado por este ion (16),(17).
- Receptores kainato: este tipo de receptores se activan por efecto del ácido kaínico o por el glutamato y, aunque en mucha menor proporción, por AMPA o NMDA. Cinco genes de dos familias codifican para formar las subunidades que se combinan heteroligoméricamente para constituir los receptores kainato. Los genes GLUR5, GLUR6 y GLUR7 hacen parte de una familia, mientras que KA1 y KA2 se encuentran en la otra. Dependiendo de la exacta composición de las subunidades que lo conforman, el canal asociado al receptor puede ser permeable al calcio o a cationes monovalentes (16), (17). Además, se ha comprobado experimentalmente que cuando las subunidades GLUR5 y GLUR6 contienen glutamina en lugar de arginina en el dominio M2, se induce una alta conductancia al calcio.
El grado de desensibilización de este tipo de receptor es lento al compararse con el del receptor AMPA. Esto permite que el uso de sustancias como el kainato o el ácido domóico (toxina que puede alcanzar al sistema nervioso después de consumir almejas que la contengan), en dosis tóxicas, al estimular el receptor, ocasionen la apertura de su canal por largo tiempo, lo cual deja ingresar grandes cantidades de sodio y agua a la neurona, que muere por sobrecarga osmótica.
Receptores metabotróficos del ácido glutámico
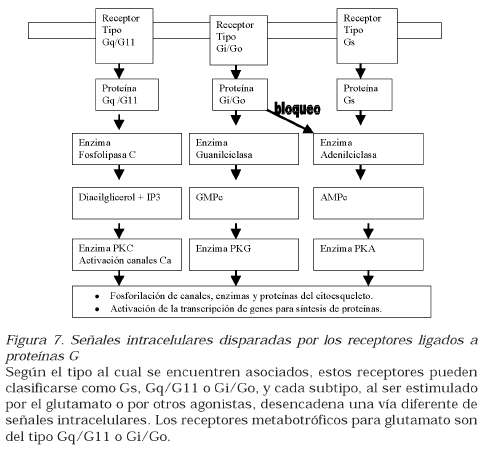
Los receptores metabotróficos del ácido glutámico son proteínas de membrana del tipo de los receptores ligados a proteínas G. Hasta el momento se han clonado ocho subtipos que se denominan mGlur 1 a mGlur 8 y su localización puede ser pre o postsináptica. Los mGlur 1 y 5 se asocian a proteínas Gq y G11, es decir, son activadores de la enzima fosfolipasa C y, por lo tanto, de la PKC y de los canales de Ca++ del retículo endoplásmico este último
por medio de la acción del inositol trifosfato (IP3). Los mGlur 2 a 8 pueden asociarse a proteínas Gi o Go (véase Figura 7). Esta interacción puede llevar, en el primer caso, a inhibir la adenilciclasa y, en el segundo, a la activación de canales de K+. Sin embargo, debe tenerse en cuenta que la función neta de estos receptores dependerá del contexto sináptico donde se encuentren, es decir, de su ubicación pre o postsináptica y del tipo de neurona en la cual estén localizados (18),(19),(2).
Homeostasis del glutamato
Relación glíaneurona
Los contactos sinápticos están circunscritos por las extensiones de los procesos de los astrocitos que sellan el compartimiento sináptico. En cada sinapsis se constituye una especie de microcámara semiaislada, cuyos límites son la membrana presináptica, la postsináptica y la envoltura glial que la rodea. Esta microestructura restringe en gran medida la libre difusión del neurotransmisor al espacio extracelular y mejora de esta forma la relación señalruido.
La envoltura sináptica también modula el pH y las concentraciones de iones, pues mantiene un gradiente electroquímico óptimo para el funcionamiento de las sinapsis. Además, al limitar el tiempo de interacción, se encarga de la mayor parte de la recaptura del neurotransmisor. Los astrocitos, cuyos procesos (como ya se mencionó) forman la envoltura glial, establecen entre sí una red constituida por uniones laxas, lo cual genera continuidad citoplasmática, que da lugar a un sincitio glial (véase Figura 8). Las uniones laxas se conocen como GAP junctions y permiten la difusión de iones como calcio, sodio, potasio y de otros osmoles como el glutamato. Por otra parte, el sincitio glial facilita la adecuada redistribución del neurotransmisor recapturado y de los iones mecanismo considerado fundamental para mantener la homeostasis del medio extracelular a pesar de las fluctuaciones impuestas por la actividad. El intercambio citoplasmático facilita la posibilidad de un búfer de potasio, la redistribución de los neurotransmisores como el glutamato y la formación de ondas de calcio, que aseguran una actividad coordinada de la red glial (21). La aparición de ondas de calcio se puede disparar por la estimulación química, eléctrica o mecánica de una célula individual, la cual redistribuye los iones hacia los astrocitos circundantes. Esta capacidad optimiza, en condiciones normales, la actividad glial y, en situaciones patológicas, crea un efecto de disolución que evita o retarda la aparición del daño.

Figura 8. Corte de corteza cerebral. Población de astrocitos marcados con el anticuerpo contra la proteína glial fibrilar (GFAP) (40X)
Se ha demostrado que las concentraciones de la proteína constitutiva de los GAP junctions, la conexina 43, se incrementa en situaciones que requieren una mayor actividad sincitial, como en modelos de epilepsia experimental inducida por ácido kaínico o en modelos de isquemia experimental. Además, se ha descrito el aumento en la señalización intercelular de calcio en muestras de tejido humano hiperexcitable (22).
Algunos datos experimentales apoyan la idea de una relación íntima entre la actividad sincronizada de neuronas glutamatérgicas y las células gliales circundantes. Por ejemplo, se ha demostrado que los astrocitos expresan receptores de AMPA y kainato y que los agonistas de estos receptores, como el ácido kaínico y el quisqualato, inducen la depolarización de la membrana de estas células. Aparte de esto, también se ha demostrado que la depolarización de los astrocitos, que utiliza agonistas como el glutamato y el quisqualato, induce elevaciones del calcio citoplasmático en otros astrocitos implicados en la misma red, lo cual se aprecia a manera de un fenómeno oscilante. También se ha visto que algunas de estas respuestas están mediadas por receptores mGluR de los tipos 1 y 5, los cuales activan la fosfolipasa C para provocar el aumento de las concentraciones de IP3 y la apertura de canales de calcio del retículo endoplásmico, que se asocia, además, a la activación de genes de expresión temprana (23).
Las concentraciones altas de glutamato extracelular son un factor importante en la generación del edema glial intracelular que acompaña a la injuria del tejido; sin embargo, cuando se da esta condición y se tiene en cuenta que el transporte de una molécula de glutamato está acompañado del ingreso de tres iones de sodio por cada molécula transportada, entonces tenemos que hay un ingreso adicional de sodio, lo que conlleva al aumento del volumen intracelular del astrocito por arrastre de agua.
En consecuencia, el incremento en el volumen de los astrocitos implica una reducción en el volumen del espacio extracelular, el cual de por sí es pequeño. Por lo tanto, las concentraciones de diferentes sustancias que estén eventualmente presentes en este medio aumentan y habiendo restricciones en su difusión, como es el caso del glutamato, se pueden generar condiciones de sobreestimulación glutamatérgica, lo cual lleva a alteraciones funcionales de la neurona (22),(23).
Como ya se ha mencionado, la principal fuente energética del sistema nervioso es el metabolismo de la glucosa; no obstante, hay indicios de una coordinación entre el ciclo de la neurotransmisión glutamatérgica y la captación glial de glucosa, que sigue una estoiquiometría 1:1. Como se explicará más adelante, la recaptura del glutamato depende del cotransporte de sodio y del contratransporte de potasio, y esto debe ser de nuevo contrabalanceado por
la bomba Na/K ATPasa, la cual requiere dos ATP para su funcionamiento, lo que obliga a la captación y glicólisis inicial de una molécula de glucosa, a fin de dejar como residuo una molécula de lactato, que pasa a la neurona para ser utilizada como fuente de energía. Esto indica que la provisión de lactato que recibe la neurona depende de la recaptura del glutamato por la glía (24).
Transportadores del ácido glutámico
En condiciones normales, las concentraciones de glutamato sináptico deben permanecer en ciertos límites que van desde 0,6 uM, en reposo, hasta 1 uM, en el momento de la actividad (25). Para mantenerlas existen varios mecanismos de control, por ejemplo: la recaptura del glutamato por las glías, las cuales se encargan de remover la mayor parte del neurotransmisor, y la recaptura por las neuronas. Una mínima parte del glutamato se difunde por fuera del espacio sináptico (26),(27),(28).
La homeostasis del glutamato en la hendidura sináptica depende principalmente de su recaptura por una familia de proteínas de membrana, denominadas transportadores de aminoácidos excitatorios (EAAT, por sus siglas en inglés, Excitatory Aminoacid Transporters), (29). Hasta ahora se han descrito cinco transportadores:
- EAAT 1 o GLAST (Glutamate/Aspartate Transporter), de localización glial, principalmente en el cerebelo (30),(31).
- EAAT 2 o GLT 1 (Glutamate Transporter 1), también glial. Es más abundante en la corteza cerebral, el estriado y el hipocampo. Se expresa también en la microglía (32),(33),(34).
- EAAT 3 o EAAC 1 (Excitatory Aminoacid Carrier 1), de localización neuronal postsináptica, principalmente en la corteza (35).
- EAAT 4, transportador neuronal presente en el cerebelo (30).
- EAAT 5, descrito tanto en neuronas como en células gliales de la retina (29).
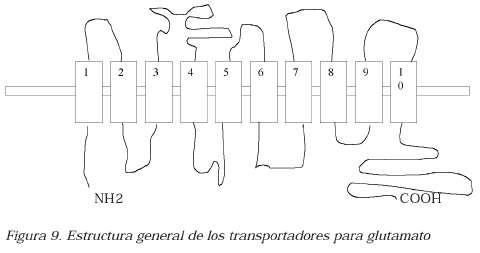
Los estudios sobre la estructura general de los transportadores los describen como moléculas proteicas de 500 o 600 residuos de aminoácidos arreglados en alfahélices, que forman entre ocho y diez segmentos transmembranales con seis segmentos en el extremo N terminal y cuatro segmentos en el extremo C terminal y loops o secciones intra y extracelulares que poseen sitios para fosforilación por PKA y PKC (Figura 9). Lo anterior indica una estrecha relación de estas moléculas con la actividad de los receptores metabotróficos para neurotransmisores, especialmente los que se consideran excitatorios, por estar ligados a proteínas G de las familias G, estimulantes de la PKA y Gq estimulantes de la PKC (36),(37),(38),(39),(40),(41).
Se ha demostrado que la función de los transportadores está bajo la influencia de los sistemas de neurotransmisores. Por ejemplo, se ha descrito que la recaptura de glutamato aumenta en la presencia de agonistas alfaadrenérgicos como la fenilefrina, mientras que la activación de betaadrenoreceptores la disminuye ligeramente (42). Adicional mente, la estimulación de los receptores para endotelinas, una familia de péptidos vasoactivos de origen endotelial con propiedades neuromoduladoras, disminuye en gran proporción la recaptura de glutamato en cultivos de astrocitos (43). La interacción entre los receptores de endotelinas y los transportadores de glutamato en los astrocitos es vital, ya que se sabe que la endotelina es liberada cuando se produce una hemorragia subaracnoidea, lo cualocasiona un aumento extracelular de glutamato por una disminución en su recaptura (44).
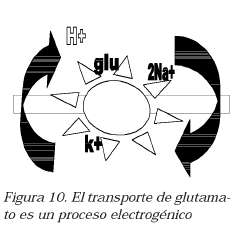
Funcionalmente, los transportadores para glutamato trabajan de acuerdo con gradientes electroquímicos, de manera que éste ingresa a la célula acompañado de dos o tres iones Na+ y sale un ion K+; no obstante, es necesaria la interacción de protones (H+), lo cual indica la importancia del pH del medio en la conservación de la actividad de los transportadores (Figura 10). Durante eventos agudos como la isquemia, las alteraciones del medio celular incluyen cambios del pH que resultan sumamente nocivos para las moléculas de membrana. En el caso de los transportadores, esta disfunción lleva a la aparición de un mecanismo de transporte reverso, en el cual el glutamato es liberado nuevamente al espacio sináptico, por lo cual se agrava el proceso excitotóxico (4 5), (4 6), (4 7).
Los transportadores tienen una distribución específica dentro del tejido nervioso. Sin embargo, en condiciones patológicas, esta distribución puede cambiar. Por ejemplo, el transportador EAAT1 de localización básicamente glial, que desempeña un papel fundamental en la regulación de la concentración máxima de glutamato en condiciones normales, en condiciones patológicas, como en isquemia o hipoxia, está presente en el compartimiento neuronal. Este hallazgo se interpreta como una adaptación del sistema para la búsqueda de una mejor regulación del glutamato en circunstancias extremas (48).
Acción glutamatérgica y plasticidad
El término plasticidad fue utilizado por primera vez por William James en 1890, según lo cita Kuno (49), quien afirmó lo siguiente:
Una conducta o hábito nuevo no es otra cosa que una nueva vía de descarga o de actividad en el cerebro [...] el tejido nervioso parece estar provisto con una extraordinaria capacidad plástica [.. .]para que el sistema sea ñexible, el tejido nervioso debe poseer una estructura suficientemente débil para ser cambiada, pero al mismo tiempo suficientemente firme para no cambiar totalmente.
Sobre la base de este principio, diferentes aproximaciones experimentales y teóricas a lo largo del siglo XX han intentado establecer indicios experimentales directos que corroboren la capacidad que tiene el sistema nervioso para ser modificado en respuesta a la actividad, a la experiencia o como consecuencia de la injuria. Vale la pena aclarar que el término plasticidad se ha utilizado en varios contextos, dependiendo del observador y de los instrumentos y métodos que se utilicen para determinarla.
Se han identificado cambios morfológicos macro y microscópicos. En el primer caso, con frecuencia asociado a cambios patológicos, se tratan de establecer cambios de volumen de la corteza cerebral, la sustancia blanca e incluso variaciones en la dimensión de los ventrículos (50),(51). En el ámbito microscópico, las pruebas de plasticidad estructural se refieren a la distribución de axones, modos de segregación, tamaños celulares, patrones de ramificación dendrítica y número de espinas. También son conocidas las adaptaciones eléctricas de ciertos conjuntos neuronales en respuesta a estimulación de alta frecuencia, como ocurre cuando se excita la vía entorrinohipocampal, que es de carácter glutamatérgico. Esta estimulación da como resultado una mayor capacidad de respuesta eléctrica que persiste por horas o incluso días; un fenómeno al que se le ha denominado potenciación a largo plazo (LTP) y se asocia a los mecanismos de aprendizaje y memoria (52),(53),(54),(55). Véase la Figura 11.
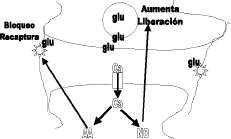
Figura 11. Potenciación a largo plazo
La entrada de calcio por activación de los receptores NMDA induce la producción de oxido nítrico (NO) y de ácido araquidónico (AA), que actúan como mensajeros retrógrados para aumentar la liberación del glutamato y disminuir su recaptura, prolongando y potenciando la actividad excitatoria del glutamato.
En consonancia con los cambios estructurales macroscópicos y las adaptaciones eléctricas de las neuronas están las adaptaciones moleculares. Éstas pueden llevar a cambios morfológicos observables o no, y pueden expresarse en las membranas celulares, el citoplasma o el núcleo. En el caso de las membranas celulares, los ejemplos más claros son los cambios en la densidad de los receptores o de sus subunidades como respuesta al tratamiento farmacológico o a la deaferentación. En el citoplasma se pueden apreciar cambios en el citoesqueleto y en las proteínas asociadas, que pueden llevar a adaptaciones estructurales sutiles como cambios en la inmunorreactividad de la proteína asociada a los microtúbulos MAP2 en zonas exofocales a un foco isquémico (56),(57) o en respuesta al tratamiento con neurolépticos. También se pueden presentar traslocaciones de proteínas del citoplasma hacia la membrana celular como es el caso de la proteinkinasa C (PKC). Bajo estas circunstancias, la membrana se hace eléctricamente más sensible fenómeno que se observa asociado a procesos de aprendizaje y memoria (58). En el núcleo se pueden presentar cambios en la expresión génica que determinan, por ejemplo, incrementos en la traducción de enzimas para la síntesis de neurotransmisores o de subunidades de receptores.
Está demostrado que múltiples sistemas de neurotransmisores inducen plasticidad. Según se ha observado en diversos modelos experimentales, las concentraciones de dopamina en la corteza frontalmedial (59) se incrementan si se somete al individuo a estímulos novedosos, lo cual está asociado a la experiencia gratificante que implica la novedad. Además, se ha comprobado que el sistema gabaérgico cortical es modificable por la actividad. Así, inmunohistoquímicamente se ha visto que la expresión de receptores GabaA y las concentraciones tanto de la enzima GAD, que sintetiza a gaba, como de ácido gamaamino butírico pueden alterarse en la corteza visual o en la corteza somatosensorial si se disminuyen los impulsos aferentes a estas zonas, lo cual tiene implicaciones sobre la extensión de los campos receptivos visual y somatosensorial (60).
Recientemente, la participación del sistema glutamatérgico en los mecanismos de plasticidad morfofuncional y, por lo tanto, del comportamiento han provocado un considerable interés, dada la influencia de este sistema sobre la estructura y dinámica de las espinas dendríticas. En el caso de la corteza cerebral, el mayor número de contactos glutamatérgicos se establecen con las espinas dendríticas sobre las cuales convergen fibras dopaminérgicas, gabaérgicasy glutamatérgicas. En el cuerpo estriado, las fibras glutamatérgicas también establecen contactos con espinas dendríticas. De igual manera, este tipo de arreglo sináptico se observa en el cerebelo, en el cual las fibras glutamatérgicas de las células granulares contactan con las espinas dendríticas de las células de Purkinje.
La morfología de las espinas dendríticas cambia en respuesta a muchos factores (aprendizaje, edad y enfermedad), procesos mediados por sistemas de señales (hormonas, factores de crecimiento), neurotransmisores (glutamato, dopamina, gaba) y elementos de la matriz extracelular (reelina). Por ejemplo, la estimulación de alta frecuencia de la vía perforante para inducir LTP en las células piramidales del hipocampo, genera cambios morfológicos en las espinas como: bifurcación, aparición de nuevas espinas, incremento en el tamaño de la sinapsis y modificaciones en la densidad postsináptica (PSD). Existen pruebas de la disminución del número de espinas dendríticas en alguno síndromes como en el de X frágil, en el de Down o en la esquizofrenia (61), (6 2), (6 3).
Las espinas maduras tienen la forma de un hongo, caracterizado por un cuello y una cabeza bulbosa, donde usualmente se observan las sinapsis. Las espinas dendríticas son compartimientos semiautónomos que, a menudo, contienen retículo endoplásmico liso y la maquinaria esencial para la traducción proteica. El principal componente del citoesqueleto de la espina es la Factina, base para explicar su continua movilidad (64).
Las propiedades estructurales de las espinas varían en sus diferentes sectores. Por ejemplo, la resistencia en el cuello de la espina es alta comparada con la de la cabeza; mientras que la alta resistencia del cuello dendrítico y su posibilidad de modificar el diámetro se considera un factor fundamental para establecer la modulación funcional de la espina (en ello participa el glutamato). Desde 1982 se ha propuesto que el diámetro del cuello de la espina puede cambiarse con ayuda de un mecanismo mediado por calcio, por activación de actina y miosina o por el desplazamiento del aparato de la espina hacia su cuello. Poco se conoce, sin embargo, sobre los mecanismos moleculares que determinan la estructura de la espina dendrítica y sus sinapsis asociadas y, particularmente, con relación a las proteínas que constituyen la PSD. En las sinapsis excitatorias, como es el caso de las glutamatérgicas, las PSD se encuentran localizadas predominantemente en las espinas dendríticas. Estas densidades postsináptica son complejos proteicos entre los que se destaca la presencia de la proteína shank, la cual se concentra en la parte profunda de la PSD.1
Como ya se mencionó, en la adaptación morfofuncional de las espinas dendríticas de las células piramidales participan varios sistemas de señalización intercelular; pero se considera fundamental la participación del glutamato, por medio de su interacción con los receptores NMDA y los metabotróficos mGluRl y mGluR5. También se señala la contribución de una proteína de la matriz extracelular, la reelina, que es producida por interneuronas gabaérgicas de las capas supragranulares de la corteza cerebral, como las células de Cajal Retzius, de doble bouquet (ramillete), multipolares y las células de Martinotti (66).
Los receptores glutamatérgicos, tanto ionotróficos como metabotróficos, establecen interacciones complejas con proteínas presentes en las densidades sinápticas, hacen enlaces con proteínas del citoesqueleto, como la actina, y, promoviendo la liberación de calcio, pueden canalizar señales hacia el retículo endoplásmico presente en las espinas (67).
Excitotoxicidad y muerte celular
La excitotoxicidad es la consecuencia de la estimulación excesiva de los aminoácidos excitatorios sobre sus receptores, lo cual lleva a una respuesta metabólica exagerada que puede ocasionar daño neuronal (68). El exceso de glutamato en la hendidura sináptica puede producirse por varios mecanismos: aumento de la exocitosis por activación de receptores presinápticos o por apertura de canales por el edema celular, exocitosis no dependiente del potencial de acción, salida no exocitótica de glutamato neuronal y liberación del glutamato por las células gliales (69).
Mecanismos de muerte celular
Una injuria puede ser tan grave que la célula rápidamente cesa su función y pierde su integridad. A este fenómeno se le conoce como oncosis, y se caracteriza por la pérdida drástica y abrupta del suplemento energético por deprivación de oxígeno y de glucosa. Téngase en cuenta que la desintegración celular permite que el contenido celular sea liberado en el tejido circundante y, como consecuencia, se induzca una respuesta inmune, inflamatoria o fagocítica, la cual puede llevar a un daño secundario en el tejido (70).
Aunque la muerte celular programada o apoptosis es un proceso natural (esencial en el desarrollo normal y la homeostasis del tejido), también se presenta acompañando algunas patologías (71). La apoptosis se caracteriza por cambios morfológicos que incluyen encogimiento celular, hinchamiento de la membrana, condensación de la cromatina y fragmentación nuclear (cariorexis). Finalmente, la célula constituye los cuerpos apotóticos, generalmente fagocitados sin el acompañamiento de una respuesta inflamatoria. El que la célula muera por oncosis o por apoptosis puede depender de si la injuria afecta la regulación de sodio, la síntesis de ATP o ambas. Cuando la disfunción del ATP es el insulto primario, las células se hinchan rápidamente debido al incremento intracelular de sodio, evento que no ocurre durante la apoptosis.
La célula muere cuando no puede mantener su equilibrio iónico (72),(73).
Todas las células poseen un mecanismo activo de muerte celular un proceso activo que involucra una serie de eventos bioquímicos intracelulares, que puede ser inducido por estímulos extrínsecos o intrínsecos (74),(75),(76). La vía final común de la apoptosis la constituye la activación de las caspasas (70),(77),(78) zimógenos de una sola cadena, que se activan por clivaje en el residuo aspartato, generando dos cadenas de enzimas activas. Éstas se pueden dividir a su vez en dos clases:
- Las que poseen un prodominio largo y autocatalizan su actividad. Entre éstas se incluyen las caspasas 8, 9 y 10, denominadas clase I o iniciadoras.
- Las que poseen prodominio corto y requieren otra enzima para su clivaje. Este grupo lo conforman las caspasas 3, 6 y 7, también conocidas como clase II o efectoras. Su activación en el tejido nervioso puede realizarse mediante tres rutas metabólicas:
- La vía de los receptores de muerte celular iniciada por medio de la caspasa inciadora 8.
- La vía mitocondrial, por medio de la caspasa inciadora 9.
- La vía de las ceramidas, mediante las caspasas iniciadoras 8 y 9.
El siguiente esquema resume las tres vías: Figura 12
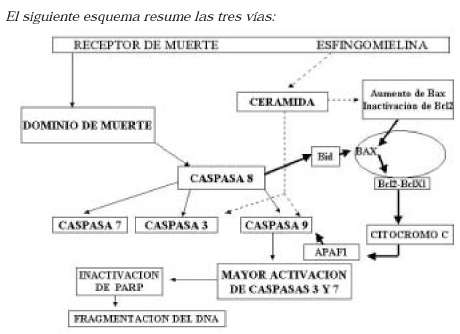
La activación de los receptores de muerte celular y el consiguiente reclutamiento de su dominio de muerte lleva a la activación de la caspasa iniciadora 8, lo cual desencadena la activación de las caspasas efectoras 3 y 7, además de otras kinasas. Las caspasas 3 y 7 inactivan a la enzima reparadora del ADN, poli ADP ribosa polimerasa (PARP), que provoca su fragmentación. En la apoptosis por la vía mitocondrial, el heterodímero antiapoptótico, formado por Bcl2 y BclXl, impide la liberación de citocromo C desde la mitocondria.
La activación de las enzimas caspasas 3 y 8 trasloca a la proteína Bid dentro de la mitocondria para estimular la traslocación de la proteína Bax a la membrana mitocondrial, la cual al formar el heterodímero con BclXl o Bcl2 crea un poro transitorio que permite la liberación de citocromo C y da paso a la formación del complejo APAF1, activador de la caspasa 9. La subsecuente activación de las caspasas efectoras culminará con la fragmentación del ADN. Por otra parte, la ruta apoptótica que utiliza la vía de las ceramidas conlleva la transformación de la esfingomielina de la membrana celular en ceramida por medio de la enzima esfingomielinasa (Smasa) normalmente éstas se convierten en otros glicolípidos de membrana como los gangliósidos. En condiciones patológicas se produce una acumulación de ceramidas, que puede activar las caspasas, aumentar la expresión de la proteína proapoptótica Bax e inactivar la antiapoptótica Be 12.
Toxicidad oxidativa por glutamato
El término excitotoxicidad hace referencia al daño neuronal inducido por el aumento en las concentraciones de calcio intracelular en respuesta a una sobreestimulación de los receptores para glutamato. Sin embargo, éste no es el único mecanismo de lesión celular. Un fenómeno paralelo, caracterizado por el aumento de radicales libres, puede presentarse en situaciones de elevación aguda o crónica del glutamato extracelular. La producción de radicales libres puede darse por la convergencia de, al menos, dos vías diferentes: la entrada de calcio a través de receptores NMDA y su liberación desde el retículo endoplásmico, lo cual induce la activación de diversas enzimas, como la calcio calmodulinakinasa (CaM Kinasa), entre cuyos sustratos se cuentan dos tipos de oxido nítrico (NO) cintazas: la nNOS (neuronal) y la eNOS (endotelial). La actividad prolongada de estas enzimas lleva a la depleción del sustrato Larginina y la producción de no es remplazada por la producción de radicales peroxinitrito (ONOO) y anión superóxido (02) (79),(80),(81).
Simultáneamente, el aumento de la concentración del glutamato en el espacio extracelular modifica la función del antiportador cisteínaglutamato, el cual exporta ácido glutámico al exterior de la célula a cambio de cisteína, necesaria para la síntesis del glutatión, el principal antioxidante de la célula, encargado de la neutralización de radicales libres (82).
Los efectos celulares de estos radicales incluyen la alteración de la fluidez de las membranas, la alteración de los sistemas de transporte mitocondrial y de la membrana plasmática y la inhibición de la enzima glutamina sintasa, a fin de impedir la conversión del glutamato en glutamina y permitir una mayor liberación al espacio extracelular, lo cual exacerba el fenómeno excitotóxico.
Pie de Página
1 La proteína shank puede desempeñar un papel importante en la interfaz entre la PSD, el citoplasma de la terminal postsináptica y el citoesqueleto. Adicionalmente, existe una interacción entre la proteína shank y la cortactina, proteína de enlace, que trasloca el citoesqueleto en respuesta a estímulos extracelulares (65).
Agradecimientos: esta publicación es parte del proyecto Evolución temporal de los cambios en la corteza cerebral inducidos por isquemia. Los autores desean expresar su agradecimiento a la Vicedecanatura de Investigaciones de la Facultad de Salud y la Vicerrectoría de Investigaciones de la Universidad del Valle, por su apoyo. Igualmente, a los miembros del Centro de Estudios Cerebrales por sus comentarios y sugerencias.
Bibliografía
1. Gallo V, Ghiani C A. Glutamate receptors in glia. New cells, new inputs and new functions. Trends in Pharmacol Sci 2000; 21:25-28. [ Links ]
2. Bergles D E, Diamond J S, Jahr C E. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol 1999; 9:29-38. [ Links ]
3. Pitts FN, McClure JN. Lactate metabolism in anxiety neurosis. New Engl Jou Med 1967; 277 1329-36. [ Links ]
4. DagerSR, Richards T, Strauss W, ArtruA. Singlevoxel HMRS investigaron on brain metabolic changes during lactateinduced panic. Psy Res 1977; 76: 89-99. [ Links ]
5.Ottersen OP, StormMatisen J. Biochemistry and anatomy of transmitter glutamate. In: Glutamate Handbookof Chemical Neuroanatomy. s. 1.: Elsevier2000; 18. p. 144. [ Links ]
6. SudhofT. Intracellulartrafficking. In: Basic Neurochemistry. Molecular, Cellular and Medical Aspects. 6thed. s. 1.: LippincottRaven; 1999. p. 175-88. [ Links ]
7. Nestler E., Hyman S, Malenka R. Biochemistry of neurottransmitter reléase. In: Molecular Neuropharmacology. A Foundation for Clinical Neuroscience. s. 1.: Me GrawHill; 2001. p. 7083. [ Links ]
8. Zenisek D, Steyer JA, Almers W. Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 2000. 406-24 [ Links ]
9. Matthews G. Vesicle fiesta at the synapse.Nature 2000. 406/24 [ Links ]
10. Mac Dermott A, Role L, Siegelbaum S. Presynaptic ionotropic receptors and the control of transmitter reléase. Ann Rev Neurosc 1999; 22: 443-85. [ Links ]
11.Miller R. Presynaptic receptors. Ann Rev of Pharmacand Tox 1998; 38: 201-27. [ Links ]
12. Reith ME. From first to fourth messengers in the brain. In: Reith MEA, editor. Cerebral Signal Transduction. From First to Fourth Messengers. Totowa: Humana Press; 2000. p. 323. [ Links ]
13. Seeburg PH, Monyer H, Sprengel R, Burnashev N. Molecular biology of NMDA receptors. In: CollingridgeGL, Waltkins JC, editors. The NMDA Receptor. 2nd ed. s. 1.: Oxford University Press; 1994. p. 147-57. [ Links ]
14. Danysza W, Parsons C. Glycine and NMDA receptors. Physiological significance and possible therapeutic applications. Pharmac Rev 1998; 50 (4): 597-664. [ Links ]
15. Collingridge G. Memories of NMDA receptors and LTP. Trends in Neurosc 1995; 12: 546. [ Links ]
16. Lerma J et al. Excitatory amino acid activa ted channels. In: Soria B, Ceña V, editors. Ion Channel Pharmacology. s. 1.: Oxford University Press; 1998. p. 399-421. [ Links ]
17. Myers S, Dingledine R, Borges K. Genetic regulation of glutamate receptor ion channels. Ann Rev of Pharmac andTox 1999; 39: 221-41. [ Links ]
18. Conn J, Pin JP. Pharmacology and function of metabotropic glutamate receptors. Ann Rev of Pharmac and Tox 1997; 37:205-37. [ Links ]
19. Krieger P, Hellgren J, Kettunnen P, JabbarA. Interactions between metabotropic and ionotropic glutamate receptors regulates neuronal network activity. The Jour Neurosc. 2000;20(14):538-291. [ Links ]
20. Cartmell J, Schoepp D. Regulation of neurotransmitter reléase by metabotropic glutamate receptors. Jou of Neuroch 2000; 75(3): 889-907. [ Links ]
21. Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Muller C et al. Neuronalglial interaction and behaviour. Neuroscand Bio Rev 2000; 24: 295-340. [ Links ]
22. Hansson E.Muyderman H, Leonova J, Aliason, L, Sinclair J, Blostrand Fetal. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamateinduced cell swelling and gapjunction communication. Neuroch ínter 2000; 37: 317-29. [ Links ]
23. CarmignotoG. Reciprocal communication systems between astrocytes and neurons. Progin Neurob 2000; 62: 561-81. [ Links ]
24. Sibson N, Dhankar A, Masón G, Rothman D, Behar K, Shulman R. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. PNAS 1998; 95:316-21. [ Links ]
25. DavenportHW. Early history of the concept of chemical transmission of the nerve impulse. Physiologist 1991; 34:129-90. [ Links ]
26. Bergles DE, Diamond J S, Jahr CE. Clearance of glutamate inside the synapse and beyond. CurrOpin Neurobiol 1999; 9(3): 29-38. [ Links ]
27. Palmada M, Centelles J. Excitatory aminoacid neurotransmision. pathways for metabolism, storage and reuptake of glutamate in the brain. Front in Biosc 1998:701-718. [ Links ]
28. Wang G , Chung H , Schnuer J , Pratt K , Zable A, Kavanough M . et al. High affinity glutamate transport in rat cortical neurons in culture. Molec Pharmac 1998 ; 53 : 88-96. [ Links ]
29. Danbolt NC. Glutamate uptake. Progin Neurob2001;65: 11-05. [ Links ]
30. Itohetal. Expression of two glutamate transporten, GLAST and EAAT 4 in the human cerebellum: their correlation in development and neonatal hipoxicischemic damage. Jouof Neuropand Exper Neur 1998; 57(6): 554-62. [ Links ]
31. González M, OrtegaA. Regulation of high affinity glutamate uptake activity in Bergmann glia cells by glutamate. Brain Res 2000; 866: 73-81. [ Links ]
32. PlachezC, Danbolt NC, RécansensM. Transient expression of the glial glutamate transporten GLAST and GLT1 in hippocampal neurons in primary culture. Jou of Neurosc Res 2000; 59: 587-93. [ Links ]
33. Mathern G, Mendoza D, LozadaA, Pretorius J, Dehenes Y, Danbolt N et al. Hippocampal GABA and glutamate transporter inmunoreactivity in patientswith temporal lobe epilepsy. Neurology 1999; 52: 453-62. [ Links ]
34. Nakajima K, TohyamaY, KohsakaS, KuriharaT. Ability of rat microglia to uptake extracellular glutamate. Neurosc Lett2001; 307: 17-14. [ Links ]
35. Conti F, De Biasi S, MinelliA, Rothestein J, Melone, M. EAAC 1, a highaffinity glutamate transporter is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cerebral Cort 1998; 8:108-16. [ Links ]
36. Seal RP, Amara SG. Excitatory amino acid transporters. Afamily in flux. Ann Revof PharmacandTox1999; 39:431-56. [ Links ]
37.Gegelavishi G, Shousboe A. High affinity glutamate transporters. Regulation of expresión and activity. Molec Pharmac 1997; 52: 615. [ Links ]
38.Tanaka K. Functions of glutamate transporters in the brain. Neurosc Res 2000; 37: 159. [ Links ]
39. Takanshi M, Billups B, Rossi D, Sarantis M, Hamman M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. Jou of Exper Bio 1997; 200 (pt2): 401-9. [ Links ]
40. Gegelashvili G, Dehenes Y, Danbolt NC, Shousboe A. The high affinity glutamate transporters GLT1, GLAST and EAAT4 are regulated via different signalling mechanisms. Neuroch ínter 2000; 37: 163-70. [ Links ]
41. Rauen T, Wiebner M. Fine tuning of glutamate uptake and degradation in glial cells. Common transcriptional regulation of GLAST andGS. Neuroch ínter 2000; 37: 179-89. [ Links ]
42. Hansson E et al. Astroglia and glutamate in physiology: aspects in glutamate transport, glutamate induced cellswelling and gapjunction communication. Neuroch ínter 2000; 37: 317-29. [ Links ]
43. Pluta RM, Book RJ, Afshar JK, Clouse K, Bacic, M, Ehreinreich H et al. Source and cause of endotelin 1 reléase into cerebrospiñal fluid after subarachnoid hemorrhage. J Neurosurg 1997; 87: 287-293. [ Links ]
44. LópezJaramillo P. Función endotelial y radicales libres. En: Bioquímica del endotelio vascular. Implicaciones fisiológicas y clínicas. 5th ed. 2001; p. 75-86. [ Links ]
45. Shlag B, Vondrasek J, Muñir M, Kalandadze A, Zelenaia O, Rothstein J et al. Regulation of the glial Na+ dependent glutamate transporters by cyclic AMP analogs in neurons. Molec Pharmac 1998; 53: 355-69. [ Links ]
46. Zhang Y, Kanner B. Two serine residues of the glutamate transporter GLT1 are crucial for coupling the f luxes of sodium and the neurotransmitter. PNAS 1999; 96:171015. [ Links ]
47. Zhang Y, Kanner B, Bendahan A, Kavanaugh M, Zarbiv R. Molecular determinant of ion selectivity of a Na+/k+ coupled rat brain glutamate transporter. PNAS 1998; 95: 751-5. [ Links ]
48.TakahashiM, Billups B, Rossi D, SarantisM, Hamann M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. J Exp Biol 2000: 401-9. [ Links ]
49. KunoM. Historical perspective of neuronal plasticity. In: Thesynapse. Function, plasticity, and neurotrophism. Oxford: Oxford Science Publications; 1995. p. 85-8. [ Links ]
50.Altshuler LL, Bartzokis G, GriederT, Curran J, Jiménez T, Leight K et al. An MRI study of temporal lobe structures in men with bipolar disorder or schizophrenia. Biol Psicol 2000; 48: 147-62. [ Links ]
51. CrespoFacorro B, Kim JJ, Andreasen NC, O' Leary DS, Magnotta V. Regional frontal abnormalities in schizophrenia. A quantitative gray matter volume and cortical surface size study. Biol Psych 2000; 48: 11-09. [ Links ]
52. EngertF, BonhoefferT. Dendriticspinechanges associated with hippocampal longterm synaptic plasticity. Nature 1999; 399: 6670. [ Links ]
53.MateticCavatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 1999; 283:19237. [ Links ]
54.Olmo N, Galarreta M, Bustamante J, Martín del Río R, Solís J. Taurineinduced synaptic potentiation. Role of calcium and interaction with LTP. Neuroph 2000; 39: 4054. [ Links ]
55. Buonomano D. Cortical plasticity. From synapses to maps. Annl Rev Neurosc 1998; 21: 149-86. [ Links ]
56. Pimienta HJ, Arango CSA, Pedroza A, Escobar MI. Respuesta neurobiológica a la lesión cerebral isquémica. Zona de infarto, zona de penumbra y regiones exofocales. Neuroc en Col 2000; 8(1): 13-25. [ Links ]
57. ShengM, Pak DTS. Ligandgated ion channel interactions with cytoskeletal and signaling proteins. Ann Rev Physiol 2000; 62: 75578. [ Links ]
58. Van der Zee A, Douma BR, Disterhoft JF, Luiten P. GProtein kinase C signaling in learning and memory. In: Maarten E A, editor, Cerebral Signal Transduction from first to fourth messengers. Reith: Humana Press; 2000. p. 73-103. [ Links ]
59.Seamans JK, Durstewitz D, Christie B R, Stevens CF, Sejnowiski J. Dopamine D1 /D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. PNAS 2001; 98(1): 30-16. [ Links ]
60. Jones EG. Cortical and subcortical contributions to activitydependent plasticity in primate somatosensory cortex. Ann Rev Neurosc 2000; 23: 137. [ Links ]
61. Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Ann Rev Neurosc 2001; 24: 299-325. [ Links ]
62. Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragüe X gene, FMR1, has characteristics of an RNA binding protein 1993; 74: 29-18. [ Links ]
63. Sherman AD, Davidson AT, Baruah S, Hegwood TS, Wazin R. Evidence of glutamatergic deficiency in schizophrenia. Neurosc Lett1991; 121: 77-80. [ Links ]
64. MatusA. Postsynaptic actin and neuronal plasticity. Curr Opin Neurobiol 1999; 9(5): 56-15. [ Links ]
65. Sala C et al. Regulation of dendritic spine morphology and synaptic function by shank andhomer. Neuron2001; 31: 115-30. [ Links ]
66. Costa et al. Dendritic spine hypoplasticity and downregulation of reelin and gabaergic tonein achizophreniavulnerability. Neurobiol of Dis 2001; 8: 723-42. [ Links ]
67. Meldolesi J. Rapidly exchanging Ca++ stores in neurons. Molecular, structural and functional properties. Progin Neurob2001; 65: 309-38. [ Links ]
68. Olney J. Excitotoxic aminoacids and neuropsychiatric disorders. Ann Rev of Pharmac and Tox 1990; 30: 47-71. [ Links ]
69.Ayala J, Sánchez JC, Palacios M, Quevedo J, Escobar MI, Cruz, S. Neuronas glutamatérgicasyexcitotoxicidad. En: Sistema nervioso. Neuroanatomía funcional, neurohistología, neurotransmisores, receptores y clínica. 2nd ed. Cali: Universidad del Valle; 1998. p. 14-39. [ Links ]
70. McDonald E, Widenback A. Mechanisms of neurotoxic injury and cell death. Neurol Clin 2000; 18(3): 525-40. [ Links ]
71. Rudin C, Thompson C. Apoptosis and Disease. Regulation and Clinical Relevance of Programmed Cell Death. Ann RevMed 1997; 48:267-281. [ Links ]
72. Zeng Y, Xu Z. Coexistence of Necrosis and Apoptosis in Rat Hippocampus Following Transient Forebrain Ischemia. Neurosc Res 2000; 37(2): 113-25. [ Links ]
73. Banasak K, Haddad G, Xia Y. Mechanisms Underllying Hipoxialnduced Neuronal Apoptosis. Progin Neurob2000; 62(3): 215-49. [ Links ]
74. Sastry P, Subba Rao K. Apoptosis and the Nervous System. Jou of Neuroch 2000; 74(1): 120. [ Links ]
75.Clarke PGH. Developmental cell death. Morphological diversity and múltiple mechanisms. Anat Embryol 1990; 181: 195-201. [ Links ]
76. Kidd V. Proteolytic activities that medíate apoptosis. Ann Rev Physiol 1998; 60: 533-73. [ Links ]
77.YamashimaT. Implication of cystein proteases calpain, cathepsine and caspase in ischemic neuronal death in primates. Prog in Neurob 2000; 62(3): 27395. [ Links ]
78. De Laurnzi V, Melino G. The little devil of death. Nature 2000; 406(13): 13-56. [ Links ]
79. Prast H, PhilippuA. Nitric oxide as a modulator of neuronal function. Prog in Neurob 2001;64:51-68. [ Links ]
80. Kiss J, Vizi S. Nitric oxide. A novel link between synaptic and nonsynaptyc transmission. Trends in Neurosc 2001; 4(4): 211-5. [ Links ]
81. Sasaki M, Dawson V, Dawson T. The no signallingin thebrain. In: ReithMEA, editor. Cerebral signal transduction. From first to fourth messengers. Totowa: Humana Press; 2000. p. 151-74. [ Links ]
82. Barger S, Basile A. Activation of microglia by secreted amyloid precursor protein evokes reléase of glutamate by cystine exchange and attenuates synaptic function. J Neurochem 2001; 76(3): 846-54. [ Links ]

