










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.32 no.1 Bogotá Jan./Mar. 2003
Artículo de revisión/actualización
Sistema glutamatérgico II: alteraciones en isquemia, alzheimer y esquizofrenia
Hernán J. Pimienta Jiménez, MSc1 Adriana M. Medina Marín, M.D.2 Martha I. Escobar Betancourth, MSc3
1Profesor titular, director del Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
2 Estudiante de Doctorado en Neurociencias, Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
3Profesora titular, investigadora del Centro de Estudios Cerebrales, Facultad de Salud, Universidad del Valle.
Resumen
Las neuronas piramidales de la corteza cerebral utilizan ácido glutámico como neurotransmisor. Diferentes tipos de aferentes convergen sobre ellas. Al mismo tiempo, estas células dan lugar a fibras eferentes que interconectan ampliamente variados sectores de la corteza, los núcleos de la base y el tálamo. La tríada de interacciones recíprocas entre neuronas glutamatérgicas, gabaérgicas y monoaminérgicas en regiones límbicas, en la neocorteza y en centros reguladores de la función cortical, como los núcleos de la base, la sustancia negra y el tálamo, se consideran importantes en las actividades motora, cognitiva y emocional. En el presente artículo se presenta una visión actualizada del sistema glutamatérgico y sus posibles implicaciones en la isquemia, la esquizofrenia y la enfermedad de Alzheimer.
Palabras clave
Ácido glutámico, isquemia, esquizofrenia, enfermedad de Alzheimer.
Abstract
Pyramidal neurons of the cerebral cortex used glutamic acid as their transmitter. Different types of afferents converge over pyramidal cells. At the same time, this cells give rise to efferent fibers that Inter.-connect widely different sectors of the cerebral cortex, basal ganglia and thalamus. The triad of reciprocal interactions between glutamatergic, gabergic and monoaminergic neurons in limbic regions, in the neocortex and in regulatory centers of cortical function such as basal ganglia, substantia nigra and thalamus are considered fundamental for the motor, cognitive and emotional activities. In the present article we offer an update review of the glutamatergic system and its possible role in ischemia, schizophrenia and Alzheimer disease.
Key Words
Glutamic acid, ischemia, schizophrenia, alzheimer disease.
Introducción
Desde el punto de vista patológico, la disfunción glutamatérgica se ha considerado un factor contribuyente en el daño inducido por isquemia, trauma, epilepsia, tumores cerebrales, diversas encefalopatías, infección por VIH, envenenamiento por mercurio y enfermedades crónicas, como la enfermedad de Alzheimer, la esclerosis lateral amiotrófica (ELA) y la corea de Huntington. También se ha sugerido una disfunción de las sinapsis glutamatérgicas en algunos circuitos telencefálicos, especialmente corticocorticales y corticoestriatales, como eventos subyacentes en enfermedades como la esquizofrenia, el trastorno bipolar con psicosis y las alteraciones de la memoria emocional, uno de cuyos factores desencadenantes puede ser el estrés crónico o agudo, según se ha demostrado en algunos modelos experimentales.
En el presente artículo se revisan los principales conceptos sobre el sistema de neuronas glutamatérgicas: localización anatómica, macrocircuitos, microcircuitos, y la implicación de estos sistemas en patologías como la isquemia, la enfermedad de Alzheimer y la esquizofrenia.
El sistema glutamatérgico cortical en el contexto de múltiples señales
Las neuronas responden a diferentes sistemas de señales, los cuales se pueden catalogar como:
- Neurotransmisores: éstos pueden inducir a excitación o a inhibición, dependiendo de su interacción con los receptores.
- Neuromoduladores: su actividad no corresponde a excitación ni a inhibición, y su efecto depende del estado funcional de sistemas de neurotransmisores, a la vez que determina cambios moleculares que influyen sobre la actividad excitatoria o inhibitoria.
- Neurohormonas (hipotalámicas y periféricas): sus efectos determinan cambios moleculares que se reflejan especialmente en la membrana celular y en algunos casos en la membrana nuclear. Esta interacción puede modificar la expresión fenotípica de la célula. En un sentido similar al anterior interaccionan los factores de crecimiento.
La acción recíproca de estos sistemas proporciona las condiciones para lograr un estado funcional óptimo y su diversidad garantiza tanto la heterogeneidad de comportamientos en el rango normal como la flexibilidad del sistema (1),(2).
Los de la membrana celular pueden responder a estos mensajes, gracias a dos tipos de receptores: los de actividad ionotrófica, que implican la apertura de canales como consecuencia de la interacción neurotransmisor- receptor, conllevan cambios eléctricos hiperpolarizantes o despolarizantes, que se pueden propagar al resto de la célula y cuya influencia temporal se mide en la escala de milisegundos. En un rango de tiempo intermedio (minutos, horas) están los de acción metabotrófica, que se caracterizan por que los receptores no constituyen canales iónicos y están acoplados tanto a sistemas de proteínas G como a una cascada de señales, cuyas acciones se extienden a los organelos (retículo endoplásmico, mitocondrias, núcleo, citoesqueleto, citoplasma y membrana celular). El resultado de estas interacciones puede llevar a adaptaciones moleculares (plasticidad), lo cual puede generar estados funcionales distintos (3).
Las neuronas también están sujetas a factores de crecimiento que, a pesar de su nombre, no restringen su acción al periodo de desarrollo y maduración neuronal. Los factores como NGF (factor de crecimiento del nervio), BDNF (factor de crecimiento derivado del encéfalo), NT3 y NT4 (factores neurotróficos), CNT (factor trófico ciliar), etc. interaccionan con receptores que se localizan en las membranas celulares y que se acoplan a sistemas de señalización, que en neuronas en desarrollo promueven su maduración y en neuronas adultas, la supervivencia. Las neuronas también están bajo la influencia de hormonas de origen hipotalámico y de glándulas endocrinas, como las suprarrenales y las gónadas. Como ejemplo de lo anterior se reconoce la influencia de los glucocorticoides, mineralocorticoides y de los estrógenos sobre conjuntos neuronales corticolímbicos, que pueden promover adaptaciones morfofuncionales cíclicas. Las más conocidas corresponden a la generación de espinas dendríticas o a alteraciones en el balance entre la proliferación neuronal y la muerte celular programada (apoptosis), como ocurre en el giro dentado del hipocampo, aun en el cerebro adulto, un hecho que se considera importante en los procesos de memoria y de aprendizaje (4),(5).
Una generalización que nos puede resultar práctica, aunque simplista, es discernir entre el concepto clásico de neurotransmisión, que se correlaciona más precisamente con las acciones ionotróficas, y el de neuromodulación, cuyo ejemplo principal es el de la acción de las monoaminas (histamina, dopamina, serotonina, norepinefrina, epinefrina) y que se relacionan más con las acciones metabotróficas (6). Sin embargo, la distinción no es tan simple, ya que, por ejemplo, el ácido gamaaminobutírico (GABA), interactúa tanto con receptores ionotróficos (GABA-A, GABA-C) como con receptores metabotróficos (GABA-B). Por su parte, el glutamato, considerado el principal neurotransmisor excitatorio en el sistema nervioso, posee importantes acciones metabotróficas y desde esta perspectiva cumpliría el papel dual de neurotransmisor de acción inmediata, mientras que sus acciones metabotróficas generarían adaptaciones similares a las inducidas por los sistemas moduladores clásicos, como las monoaminas, los péptidos y las hormonas (7),(8).
Las interacciones glutamatérgicas, dada su amplia distribución, constituyen un eje fundamental en la tarea de entender fenómenos muy complejos, como las funciones mentales superiores (9),(10). Las células glutamatérgicas establecen vínculos recíprocos, promueven la actividad de células inhibitorias, están bajo la influencia de sistemas monominérgicos y colinérgicos y, además, promueven acciones sobre estos mismos conjuntos neuronales (11),(12), así como se puede ver en la Figura 1.
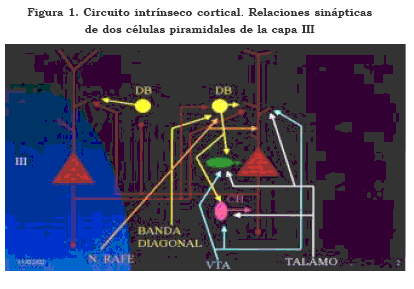
En la figura anterior se puede observar que cada célula piramidal (roja) genera un axón descendente, que puede conformar un circuito corticocortical o comisural, cuya influencia se expande a otros módulos corticales. Cada célula de este tipo genera ramas recurrentes glutamatérgicas excitatorias que las conecta recíprocamente. Además, se observa que cada axón recurrente se conecta a una célula gabérgica de doble bouquet (DB). Esta última, a la vez que inhibe a las células piramidales, inhibe a la célula en cesta (CC) y en chandelier (CH), la cual, a su vez, inhibe a la célula piramidal. Por lo tanto, en el contexto del circuito, esta célula es inhibitoria sobre la célula piramidal y al mismo tiempo desinhibitoria por su acción sobre las CC y CH, cuyos axones inhibitorios terminan respectivamente en el soma y en el segmento inicial del axón de la célula piramidal.
Se puede observar que este conjunto de neuronas está bajo la influencia de otros sistemas aferentes, cuya acción excitatoria o moduladora sobre los diferentes tipos de células agrega complejidad, pero se considera fundamental para la flexibilidad del circuito. Las aferentes serotoninérgicas proceden de los núcleos del rafé y terminan sobre las células de doble bouquet y sobre la dendrita apical de la célula piramidal. Las aferentes gabérgicas proceden de la banda diagonal de Broca y terminan sobre la célula de doble bouquet. Las aferentes dopaminérgicas proceden de la VTA y terminan sobre las células en cesta, en chandelier y sobre las espinas de las células piramidales. Las aferentes glutamatérgicas de origen talámico terminan sobre las células en cesta, chandelier y las espinas de las células piramidales.
Por simplicidad sólo se señalan algunas aferentes y se ejemplifican en el conjunto de neuronas localizadas a la derecha, pero se presume idéntico para el conjunto de neuronas de la izquierda.
La tríada de interacciones entre neuronas glutamatérgicas, neuronas gabérgicas e inervación monoaminérgica en las regiones límbicas, en la neocorteza y en centros de regulación de la actividad cortical, como el núcleo caudado, el putamen, el núcleo acumbens, la sustancia negra y la VTA, constituyen la base de las funciones motora, emocional y cognitiva (13),(14), así como se puede apreciar en la Figura 2. El sistema de neuronas glutamatérgicas y sus interacciones son de fundamental importancia en la búsqueda de los mecanismos patofisiológicos de las diferentes entidades neurológicas y de las enfermedades mentales.
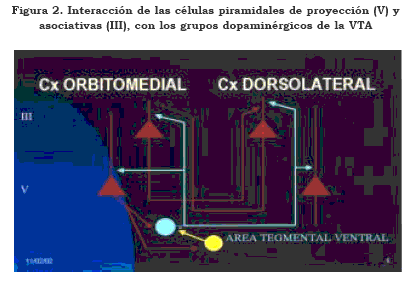
La importancia del sistema glutamatérgico parece obvia cuando observamos las siguientes cifras comparativas: el 70% de las neuronas corticales utilizan glutamato como neurotransmisor. Si, como lo señala Szentagothai (15), cada módulo cortical posee dos mil células piramidales aproximadamente y en la corteza cerebral se calculan tres millones de módulos, el número de células piramidales alcanzaría los seis mil millones. Se calcula que entre el 25%-30% (16),(17) de las células corticales son gabérgicas. Esto daría aproximadamente 2.500 millones. Debe recordarse que algunas coexpresan además de GABA, neuropéptidos.
En la Figura 2 se indican dos módulos corticales hipotéticos, uno localizado en la corteza orbitomedial, relacionada con la función emocional, y el otro en la corteza dorsolateral, relacionado con las funciones cognitivas. Las neuronas de estos dos módulos están conectadas recíprocamente. Por simplicidad no se indican las conexiones con las neuronas inhibitorias que se señalaron en la Figura 1. De la lámina V del módulo orbitomedial se proyectan axones glutamatérgicos que conectan simultáneamente a neuronas gabérgicas (amarillas) y a neuronas dopaminérgicas (azul) de la VTA, cuyo axón asciende y conecta las neuronas piramidales de las capas III y V de ambos módulos. La reciprocidad de las interacciones entre la corteza cerebral y los grupos monoaminérgicos en general y en particular los dopaminérgicos demuestran la influencia que la actividad cortical encargada de la función consciente puede ejercer sobre sistemas considerados clásicamente moduladores de la actividad cortical. La inervación extrínseca a la corteza cerebral, aquella que procede de neuronas cuyos cuerpos están por fuera de la corteza, proviene fundamentalmente del tálamo y son glutamatérgicas. Su número no se ha calculado, pero no alcanza a la decena de millones.
La inervación colinérgica, que procede de los núcleos basales de Meynert, es inferior al millón de células. Las neuronas dopaminérgicas que se proyectan a la corteza cerebral desde la VTA, representan una proporción de algunos cientos de miles. Igual sucede con los grupos serotoninérgicos del rafé y del locus coeruleus, cuyas células noradrenérgicas no alcanzan a cien mil. Los grupos histaminérgicos del hipotálamo (región tuberomamilar) contienen algunos miles de células, cuyos axones divergen a las diferentes regiones corticales. Finalmente, existen algunos grupos de neuronas gabérgicas de la banda diagonal de Broca y de la VTA, que se proyectan a la corteza cerebral y cuyo número también es reducido.
Puede ser de utilidad apreciar entonces que en la corteza cerebral existen al menos dos patrones básicos de inervación:
- Uno mediado por interacciones glutamatérgicas corticocorticales y talamocorticales específicas, con un sistema axonal circunscrito, topográficamente organizado y modular, lo cual garantiza especificidad. Dentro de estas interacciones debemos considerar las relaciones entre las células glutamatérgicas, entre células glutamatérgicas y gabérgicas y viceversa (18),(19).
- El otro sistema de inervación cortical incluye las aferentes talámicas inespecíficas y los sistemas monoaminérgicos. En éstos no es obvia una representación topográfica, dado que los axones están muy ramificados y una sola fibra aferente diverge hacia múltiples sitios. Esta organización se compensa por el escaso número de células de origen, pero no son un arreglo óptimo para garantizar especificidad. Sin embargo, su impacto sobre el sistema específico de neuronas glutamatérgicas y gabérgicas es determinante, pues su disfunción está asociada a numerosas alteraciones del comportamiento (20),(21). De acuerdo con lo anterior, al ser el sistema de neuronas piramidales glutamatérgicas corticales el punto de convergencia de múltiples influencias electroquímicas y, al mismo tiempo, el origen de sistemas de comunicación inter e intracorticales y con estructuras subcorticales, es importante para el psiquiatra, y en general para los profesionales que estudian el comportamiento normal y patológico, adquirir algunos principios fundamentales sobre el sistema glutamatérgico, a fin de comprender los niveles de sistemas de neuronas, macrocircuitos, microcircuitos y organización sináptica.
Circuitos glutamatérgicos
En la Figura 3 se muestran de manera esquemática la localización de las neuronas glutamatérgicas, sus proyecciones y sus interacciones con otros sistemas de neuronas
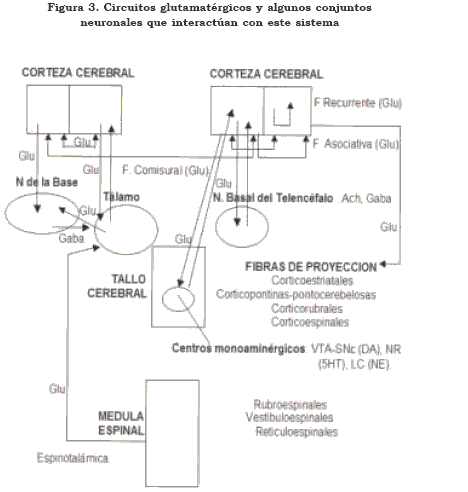
En la Figura 3 se observa que en la corteza los circuitos corticocorticales ipsilaterales y comisurales son recíprocos y mediados por el glutamato y que constituyen la base de los mecanismos de integración inter e intracorticales. Se nota que las relaciones corticotalámicas y talamocorticales son recíprocas y de carácter glutamatérgico, mientras que las proyecciones talamoestriatales y corticoestriatales, aunque son glutamatérgicas, no son recíprocas. La corteza cerebral proyecta axones glutamatérgicos al estriado (caudado-putamen), pero la influencia de éstos sobre la corteza es indirecta a través del tálamo, vía globo pálido y sustancia negra reticulada y mediada por el ácido gamaaminobutírico. Las fibras corticales de proyección se extienden a diferentes conjuntos neuronales en el tallo cerebral y en la médula espinal. Observe que algunos grupos de neuronas del tallo cerebral (rubroespinales, retículoespinales, vestíbuloespinales, pontocerebelosas) también son glutamatérgicas. Aunque de manera tradicional se han resaltado las proyecciones corticopetales de los conjuntos neuronales noradrenérgicos, serotoninérgicos,dopaminérgicos y colinérgicos, dada su influencia sobre la actividad cortical en todos sus aspectos, motor, sensorial, cognitivo y emocional, es importante considerar la influencia que los sistemas glutamatérgicos de origen cortical ejercen sobre estos conjuntos neuronales.
En la corteza cerebral, el glutamato es el neurotransmisor de las células piramidales, las cuales representan dos tercios de las neuronas corticales. Las proyecciones glutamatérgicas que se derivan de la corteza cerebral, se extienden a estructuras moduladoras de la función cortical como los núcleos de la base, el tálamo y grupos monoaminérgicos y colinérgicos (22),(23),(24),(25). Los núcleos de la base no poseen neuronas glutamatérgicas; sin embargo, reciben un aporte notable de fibras glutamatérgicas procedentes de la corteza cerebral y del tálamo. Estas proyecciones forman parte de los circuitos paralelos cortico-estriado-tálamocorticales, los cuales se consideran indispensables no sólo para la regulación motora, sino para la emocional y cognitiva. Se ha sugerido que la disfunción de estos circuitos puede estar asociada a algunos de los síntomas que acompañan a entidades como la esquizofrenia, el TOC y el trastorno bipolar (26),(27)
Los sistemas de interacción talamocortical y corticotalámico están mediados por circuitos glutamatérgicos. A las ya conocidas proyecciones talamocorticales hacia las áreas corticales sensoriales, hay que agregar la transferencia desde el tálamo hacia la corteza de señales motoras, asociativas y límbicas. Éstas involucran sectores multimodales de la corteza occipital, parietal, temporal y, especialmente, de las regiones prefrontales. Estas últimas están vinculadas con las funciones mentales superiores, los procesos de atención, la inhibición de la conducta, la memoria operativa, la motivación y el estado de ánimo (28),(29).
Neuronas glutamatérgicas de la corteza cerebral
Como ya se mencionó, en la corteza cerebral —incluido el hipocampo— la mayoría de las neuronas y de los circuitos son glutamatérgicos. Las neuronas glutamatérgicas de la corteza cerebral son células de forma piramidal y están presentes en todas las láminas de la neocorteza, excepto la I, pero predominan en las III, V y VI. Su tamaño fluctúa entre pequeñas (alrededor de 12 mm), medianas (30 mm) y grandes (hasta 100 mm). Se caracterizan, además, por la presencia de espinas en las dendritas basales y apical (Figura 4). Su axón es descendente y provee cerca de su inicio colaterales recurrentes que, en general, hacen contacto sináptico con neuronas piramidales y no piramidales (gabérgicas), localizadas en el mismo módulo o en módulos adyacentes (30),(31). Las ramas principales se extienden a otras áreas corticales en el mismo hemisferio o en el contralateral y hacen contacto en su gran mayoría con otras neuronas piramidales —alrededor del 90%— (Figura 5). Es importante señalar que estas fibras también contactan interneuronas gabérgicas, las cuales forman circuitos locales intracorticales (32),(33).
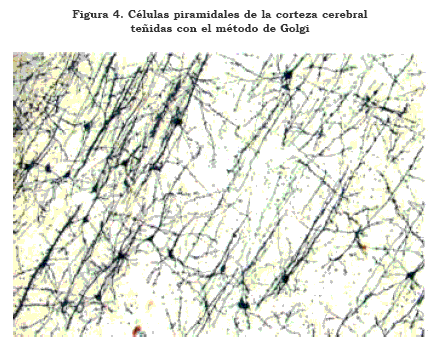
En la figura anterior se pueden observar células piramidales de las capas III y V. Se identifican las dendritas apicales, basales y espinas dendríticas. Observe que las células presentan su axón descendente, en el caso de la lámina III, lo cual da lugar a fibras comisurales o asociativas y, en el caso de la lámina V, a fibras de proyección. Note la presencia de axones recurrentes, los cuales se consideran fundamentales para el procesamiento intracortical, especialmente la memoria operativa. Según Lewis (89), estas recurrentes estarían más restringidas en la corteza prefrontal de pacientes esquizofrénicos (25X).
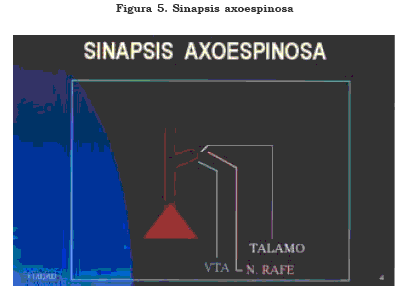
Las interacciones entre neuronas glutamatérgicas (piramidal- piramidal), entre glutamatérgicas (piramidales) y gabérgicas (interneuronas), que a su vez conectan a otras interneuronas y a otras células piramidales, constituyen los principales circuitos que subyacen a la función cortical. Este sistema de interacciones excitatorias, inhibitorias y desinhibitorias está a su vez regulado por sistemas de fibras noradrenérgicas (locus coeruleus), dopaminérgicas (VTA, sustancia negra compacta), serotoninérgicas (núcleos del rafé), histaminérgicas (región tuberomamilar), gabérgicas (banda diagonal de Broca, VTA y región septal) y peptidérgicas, principalmente de origen intrínseco (4),(35), así como se puede ver en la Figura 5.
En la figura anterior las espinas de las células piramidales son unidades integrativas muy complejas, sobre las cuales se han demostrado varios ejemplos de plasticidad morfofuncional. Las interacciones glutamatérgicas de las células piramidales se realizan en una elevada proporción sobre las espinas. Estas aferentes pueden proceder de la misma corteza o del tálamo. Además, se han identificado otras aferentes a la espina, que se consideran moduladoras como las dopaminérgicas de la VTA y las serotoninérgicas de los núcleos del rafé.
Patologías asociadas con el deterioro de la función glutamatérgica
La disfunción del sistema glutamatérgico puede expresarse como un aumento o una disminución de su concentración en el medio extracelular. En el primer caso se produce una sobreestimulación de sus receptores. En el segundo hay un descenso de la actividad excitatoria, lo cual, a largo plazo, puede alterar la función normal del sistema nervioso, en particular de algunos circuitos de la corteza cerebral y de sus proyecciones. Se ha demostrado el papel del incremento en la actividad glutamatérgica en entidades de inicio agudo como la isquemia cerebral y de tipo crónico, como la enfermedad de Alzheimer. También se ha propuesto hipoactividad del sistema glutamatérgico en procesos de fisiopatología compleja y aún no esclarecida, como la esquizofrenia.
Isquemia
La isquemia cerebral es una de las principales causas de morbimortalidad y discapacidad física y mental en el mundo. En Estados Unidos se ha calculado que alrededor del 31% de los sobrevivientes requieren asistencia en su autocuidado, el 20% necesita ayuda para movilizarse, el 71% presenta disminución de su capacidad laboral cuando son evaluados siete años después del evento isquémico y el 16% debe permanecer recluido en instituciones neurológicas o psiquiátricas, debido a la aparición de síndromes clínicos como depresión, manía, trastorno bipolar o trastornos de ansiedad posteriores al evento isquémico (36),(37),(38).
Diversos fenómenos fisiopatológicos se han descrito en los diferentes periodos que siguen al evento agudo. Entre ellos se cuentan: la disminución de la disponibilidad de moléculas de alta energía como el ATP, la hipoperfusión capilar del área lesionada, la inhibición de la síntesis proteica durante el periodo de reperfusión, la peroxidación de lípidos de las membranas celulares por radicales libres, las alteraciones de la fosforilación de proteínas del cito esqueleto y la presencia de cambios morfológicos evidentes tanto en la histoquímica como en las histopatologías convencionales (36). Todos estos eventos se encuentran ligados directa o indirectamente al estado funcional de la neurotransmisión glutamatérgica.
La disminución de fosfatos de alta energía implica la existencia de alteraciones en el metabolismo de la glucosa, pues el ciclo de captación y de utilización de glucosa en el sistema nervioso central está íntimamente ligado a la homeostasis del ácido glutámico. En condiciones normales los astrositos captan un elevado porcentaje de la glucosa sérica, para convertirla en ácido láctico, uno de los sustratos energéticos de la neurona (39). No obstante, la velocidad de captación de la glucosa por el astrocito está modulada por la velocidad de recaptura del glutamato sináptico y por los transportadores gliales, como GLAST y GLT1, los cuales trabajan con un gradiente de sodio, de manera que por cada molécula de glutamato transportada, ingresan tres iones de sodio al interior de la célula (40), (41),(42),(43).
Esta entrada de sodio debe ser contrabalanceada por la bomba Na/K ATPasa, la cual requiere dos ATP para cada ciclo funcional. Éstos se obtienen de la glucólisis inicial de la glucosa a ácido láctico, y se ha comprobado la existencia de una estequiometría 1:1 entre la recaptura del glutamato extracelular y la captación de glucosa (44),(45). Así, el incremento de la neurotransmisión excitatoria glutamatérgica induce el aumento de la captación de glucosa por los astrocitos y permite una mayor provisión de lactato como sustrato energético para las neuronas (46).
El ácido láctico tiene un reconocido efecto vasodilatador, por lo que se espera que exista un aumento del flujo sanguíneo local en respuesta a la liberación sináptica de glutamato. Sin embargo, durante el fenómeno isquémico, la excesiva producción y exportación de ácido láctico por los astrocitos como consecuencia tanto de la excitotoxicidad como de la disminución del suministro de oxígeno, facilita la inducción de una vasodilatación intensa, que podría acarrear una pérdida de la presión de perfusión tisular acompañada de edema extracelular. También es conocido el efecto ansiogénico del ácido láctico, el cual ha sido demostrado en modelos de pánico inducido por lactato y en trastorno de ansiedad, lo cual puede agravar las manifestaciones clínicas del evento isquémico (47),(48).
La disminución de la síntesis de proteínas es un problema multifactorial, pero puede atribuirse en parte a la estimulación de los receptores metabotróficos para glutamato de tipo mGluR 2 al 4 y 6 al 8, los cuales son receptores ligados a proteínas G de tipo inhibitorio como Gi y Go, por lo que la activación de los receptores se asocia a la inhibición de la enzima adenilciclasa y a la disminución de segundos mensajeros intracelulares, como el AMP cíclico, que son parte de las cascadas de señales intracelulares necesarias para la trascripción de genes y, por lo tanto, para la síntesis proteica (49),(50).
El daño de las membranas celulares por la presencia de radicales libres depende en parte de la producción de radicales derivados del óxido nítrico por activación excesiva de las enzimas óxido nitricosintasa dependientes de calcio, lo cual ocurre en estados patológicos asociados a elevaciones del calcio intracelular, como lo es la excitotoxicidad por glutamato (51). Por otra parte, el exceso de glutamato extracelular altera la función del antiportador glutamato/cisteína. Este último sustrato es esencial para la producción de glutatión, el principal neutralizador de radicales libres de la célula.
Los eventos agudos como la isquemia y el trauma crean cambios en el microambiente celular, que incluyen alteraciones del pH, alteración de los gradientes electroquímicos de la membrana celular, edema celular, apertura de canales y pérdida del equilibrio iónico (52),(53). Se ha demostrado que en estos eventos hay un aumento de la exocitosis de glutamato por las terminales sinápticas, liberación de glutamato glial sensible a indometazina (es decir, posiblemente dependiente de prostaglandinas), y liberación del glutamato neuronal por función reversa de los transportadores. Sin embargo, el transportador glial persiste en su actividad hasta que la reducción energética de la célula sea muy grave. En estos casos se ha detectado una disminución de la expresión del transportador GLT1 (54),( 55), (56), (57), (58), (59), (60), (61), (62), (63).
Enfermedad de Alzheimer: interacción glutamato-acetilcolina
Tradicionalmente la aparición de la sintomatología propia de esta entidad se ha atribuido a la alteración de la función del sistema colinérgico, a la acumulación del péptido betamieloide y a la degeneración progresiva de grupos neuronales, que lleva a la formación de los ovillos neurofibrilares y neuritas anormales, que son el hallazgo histopatológico característico de la enfermedad.
Los núcleos productores de acetilcolina, como el núcleo basal de Meynert y el núcleo septal medial, están afectados preferencialmente. Lo cual explica en parte la respuesta positiva de los pacientes al manejo con fármacos colinérgicos en los estadios iniciales de la enfermedad. La alteración en la función de diversos sistemas de neurotransmisores, los cuales incluyen acetilcolina, noradrenalina, serotonina y glutamato, se explica en parte por la deaferentación resultante de la degeneración neuronal de algunos núcleos subcorticales monoaminérgicos y colinérgicos y en regiones de la corteza cerebral como el hipocampo y la corteza entorrinal (en el lóbulo temporal) y en sectores del lóbulo frontal (64),(65),(66). Sin embargo, la pérdida de dichas aferencias no es la única causa de la disfunción de esos sistemas de neuronas. Los cambios del citoesqueleto celular pueden estar presentes aun cuando no se evidencian cambios en la histopatología convencional. La fosforilación anormal de proteínas asociadas al citoesqueleto, como la proteína Tau, ocasiona la pérdida de la estabilidad de los microtúbulos y otros filamentos. Éstos pierden no sólo su morfología, sino su funcionalidad, lo que impide el acople adecuado de los receptores metabotróficos con sus sistemas de transducción de señales, lo cual dificulta la generación de una respuesta neuronal adecuada aun en presencia del neurotransmisor (67),(68). El péptido betamieloide se produce por el clivaje inadecuado de su proteína precursora (APP). El procesamiento no amiloidógeno de la proteína APP es modulado por la enzima PKC, la cual se activa en respuesta a la estimulación de receptores metabotróficos del tipo Gq y G11. Esta modulación se ha comprobado experimentalmente para los receptores muscarínicos M1 y M3 y para los receptores metabotróficos para glutamato —mGluR 1 y 5— (64),(69).
Existe abundante información referente a la pérdida de la homeostasis del calcio en la enfermedad de Alzheimer, por lo que se considera que parte de la compleja patofisiología de esta enfermedad puede deberse a fenómenos de excitotoxicidad crónica.
La elevación de productos derivados de la activación de la enzima calpaina, una proteasa dependiente de calcio, fue reportada por Saito (70) en muestras de pacientes con Alzheimer. La calpaina regula diversos aspectos de la señalización intracelular, lo cual incluye la actividad de proteincinasas y proteinfosfatasas, que modifican el estado de fosforilación de proteínas asociadas al citoesqueleto. Sus cambios morfológicos en esta enfermedad son bien conocidos, por ejemplo, los ovillos neurofibrilares y neuritas características de la histopatología se atribuyen a la hiperfosforilación de la proteína Tau, que tiende entonces a organizarse en filamentos helicoidales pareados (64),(71),(72). Cambios similares a los de estos pacientes en la inmuno reactividad a la proteína Tau fueron reportados por Mattson (73) en neuronas de hipocampo sometidas a toxicidad por glutamato durante una hora. Las alteraciones del citoesqueleto y de la actividad de enzimas cinasas como la PKC se asocian a los déficits cognitivos de memoria y a la desorientación características de la entidad. La enzima PKC es activada durante la estimulación de receptores ligados a proteínas G de los tipos Gq y G11, como lo son los receptores muscarínicos M1 y M3 y los metabotróficos para glutamato mGluR 1 y 5. Se ha observado, en modelos experimentales, un aumento de la activación de la PKC en el hipocampo después de procesos de aprendizaje espacial. Además, la utilización de inhibidores experimentales de las cinasas impide la consolidación de la memoria en estas pruebas. Por lo tanto, las alteraciones de la memoria, la desorientación y la deficiencia cognitiva en estos pacientes pueden estar relacionadas con la interrupción de la transducción de señales que normalmente ocurre en respuesta a los neurotransmisores como el glutamato y la acetilcolina (74).
Dentro de los fenómenos fisiopatológicos de la enfermedad de Alzheimer se cuenta la inhibición de la actividad de los transportadores para glutamato y glucosa por los radicales libres inducidos por el péptido betamieloide. Las concentraciones de calcio intracelular se encuentran elevados en células que están expuestas al péptido por cuatro horas (54). Este fenómeno se debe tanto al ingreso de calcio por activación de los receptores NMDA como a la liberación de calcio del retículo endoplásmico por activación de los receptores metabotróficos de tipo mGluR1 y 5. En ambos casos la respuesta se incrementa por la mayor disponibilidad de glutamato, secundaria a la inactivación de los transportadores, por lo que es más evidente en células que expresen formas mutantes de la proteína reticular presenilina 1, como se observa en las formas autosómicas dominantes de la enfermedad (75), (76), (77), (78).
Tanto en las placas neuríticas como en las placas amielioideas hay presencia de microglías, como manifestación de un proceso inflamatorio crónico (79). Las microglías pueden generar cantidades tóxicas de glutamato por la acción del péptido betaamieloide o por su proteína precursora secretada (sAPP). Para explicar este fenómeno se ha propuesto una alteración en el antiportador glutamato/cisteína, el cual normalmente introduce glutamato a la célula a cambio de cisteína, pero desde estas circunstancias su función es inversa. Sin embargo, dado que el péptido betaamieloide influye negativamente en la actividad del transportador GLT1, por ser el principal captador de glutamato, su bloqueo, alteración o reversión podrían ser otra posibilidad que se debe considerar como factor contribuyente en el fenómeno excitotóxico mediado por la microglía. Fundamentados en lo anterior, se ha intentado contrarrestar la acción de la proteína betaamieloide sobre el transportador GLT1 con el uso de betaestradiol, pero los resultados no han sido alentadores. El panorama es más complejo si se considera que existen variedades anómalas del transportador GLT1 en enfermedades como Alzheimer y ELA (54).
Esquizofrenia
En las patologías mencionadas (isquemia y Alzheimer) se puede establecer una correlación entre hiperactividad del sistema glutamatérgico y la lesión excitotóxica, como factor contribuyente para explicar sus cambios patológicos característicos. También se establece que la causa de muerte celular, dependiendo de la intensidad del daño, puede ser necrótica o apoptótica (80). En el primer caso con secuelas histopatológicas notables, cuya característica principal es pérdida neuronal acompañada de gliosis y en las fases agudas presencia de células de la línea inmune. En el segundo caso se presenta pérdida neuronal sin gliosis o proceso inflamatorio acompañante (81).
En el caso de la esquizofrenia, en varios estudios se ha reportado la pérdida de diversos tipos neuronales en diferentes regiones, especialmente en las láminas II y VI de la corteza prefrontal y en el giro cíngulo (82),(83),(84), sin cicatrización glial, lo cual sugiere que la eliminación selectiva de algunos tipos neuronales obedece a un proceso apoptótico que, por sus características, no deja secuelas obvias en el tejido circundante. Los interrogantes por resolver son: ¿cuándo ocurre esta pérdida neuronal? ¿Se restringe al desarrollo fetal —fase crítica segundo trimestre— (85),(86), a la fase perinatal, a la infancia o a la adolescencia? ¿Existe un proceso masivo de muerte neuronal con un límite en el tiempo o es un evento progresivo que se extiende hasta la adolescencia?
Durante el desarrollo la muerte celular programada o apoptótica es fundamental que se establezca la organización citoarquitectónica y neuroquímica normal; sin embargo, desviaciones de este proceso por fenómenos de origen multifactorial genéticos o epigenéticos pueden llevar a pérdida selectiva de conjuntos neuronales. Entre los factores que pueden contribuir están la suplencia inadecuada de factores tróficos NGF, BDNF, NT3, NT4; la expresión aumentada de receptores de muerte, como p75, cuya estimulación es importante para desencadenar la cascada de muerte mediante la procaspasa 8 y la citocromo c, y la acumulación de ceramidas, que lleva a un incremento en la actividad de genes proapoptóticos como Bax y a la inactivación de genes antiapoptóticos como Bcl2. También se ha involucrado la estimulación sostenida de concentraciones moderadamente altas de glutamato, que llevarían a cantidades citoplasmáticas crónicamente elevadas de calcio, cambios de la permeabilidad mitocondrial y desencadenamiento de mecanismos de apoptosis (80). Sin embargo, no es claro todavía si existe una pérdida selectiva progresiva de tipo apoptótico, que se expanda más allá del periodo de desarrollo en regiones límbicas y neocorticales y que se pueda correlacionar en el tiempo con la aparición de los síntomas de la esquizofrenia.
Desde las primeras observaciones de Ingvar (87), quien describió que el flujo sanguíneo cerebral en la corteza prefrontal era inferior cuando se compara con el de regiones posteriores de los hemisferios cerebrales, se estableció el concepto de la hipofrontalidad para explicar algunos de los síntomas típicos de la esquizofrenia. Este hallazgo fue un importante soporte para apoyar el concepto de una disfunción prefrontal en esquizofrenia, derivado de las pruebas neuropsicológicas y del estudio de pacientes neurológicos con lesiones prefrontales, algunos de cuyos síntomas pueden compararse con los de la esquizofrenia. Posteriormente, muchos grupos de investigación, que utilizaron técnicas imagenológicas más precisas, llegaron a conclusiones similares. Sin embargo, se presentaron muchas discrepancias a medida que los métodos se fueron refinando y según algunos acontecimientos incluso se llegó a la conclusión opuesta, es decir, a sugerir incrementos en la actividad prefrontal (hiperfrontalidad), en pacientes con síntomas agudos.
Según lo señala Ebmeier (88), las discrepancias entre hiper e hipofrontalidad, debida al tratamiento con neurolépticos, se resuelven en la mayoría de los casos si se evalúa la función frontal no en estado basal, sino cuando se solicita al paciente la ejecución de una tarea cognitiva, especialmente pruebas de memoria operativa. En estos casos existe una clara tendencia a la hipofrontalidad.
¿Cuáles son las pruebas que sustentan la disfunción glutamatérgica prefrontal en esquizofrenia? Existen muchas líneas y grupos de investigación orientados a establecer las bases celulares y subcelulares de la disfunción prefrontal en esquizofrenia (89). Se ha sugerido que la disminución del volumen cortical (90),(91),(92),(93), que se aprecia en pacientes esquizofrénicos, es una consecuencia de la disminución en el neuropilo, el cual representa prolongaciones dendríticas, espinas, terminaciones axónicas corticocorticales, axones recurrentes intracorticales, aferentes talamocorticales y otros sistemas de origen extracortical como las fibras monoaminérgicas (dopamina, serotonina, norepinefrina, epinefrina), colinérgicas e incluso gabérgicas—. A pesar de esta riqueza y diversidad neuroquímica en el neuropilo, la mayoría de las relaciones sinápticas entre células piramidales y entre células piramidales y células gabérgicas son de tipo excitatorio y utilizan glutamato como neurotransmisor (93). Si existe reducción en el neuropilo, estos circuitos excitatorios podrían ser uno de los principales contribuyentes para explicar los cambios. Sin embargo, las pruebas para esto no son directas.
Lewis (89) señala que las células piramidales de la lámina III en la corteza prefrontal dorsolateral de pacientes esquizofrénicos son de menor tamaño comparado con sujetos normales. Estas células son glutamatérgicas y son la principal fuente de circuitos recíprocos corticocorticales intrafrontales y con zonas asociativas temporales y parietales, los cuales se consideran fundamentales para el reclutamiento de información relevante para la memoria operativa (working memory), función que está afectada en pacientes esquizofrénicos. Es necesario anotar que una disfunción en el sistema glutamatérgico puede desencadenar ajustes en otros sistemas de neuronas no glutamatérgicas, específicamente interneuronas gabérgicas. Su efecto puede reflejarse también en otros sistemas de fibras aferentes a la corteza. Existen indicios de disminución en la densidad de los contactos axoaxónicos sobre las células piramidales especialmente de la capa III, procedentes de las células gabérgicas de tipo chandelier (94). Sin embargo, no existen pruebas morfológicas de alteraciones en los sistemas de inervación inespecíficos de la corteza. Esto no excluye que su regulación sináptica pueda estar alterada, dada la estrecha relación que existe entre la sinapsis glutamatérgica y la inervación dopaminérgica de los mismos contactos.
Los datos para implicar la disfunción del sistema glutamatérgico en la corteza prefrontal a nivel sináptico y molecular parte del uso de antagonistas del receptor NMDA, como la fenciclidina, cuya utilización en sujetos humanos normales induce síntomas parecidos a los de pacientes esquizofrénicos (95). Se han reportado (96), (97), (98), (99) incrementos en el enlace de receptores NMDA en la corteza prefrontal de sujetos esquizofrénicos (up regulation). Igualmente, se han obtenido datos similares con respecto a los receptores tipo kainato y mGluR. Los anteriores hallazgos se han interpretado como un mecanismo compensatorio que equilibraría la disminución relativa de glutamato en estas regiones de la corteza prefrontal. Los datos anteriores ganan significancia con el análisis de la regulación por lo bajo (down regulation) en la expresión del transportador EAAT2 en la corteza prefrontal, que es el encargado de la recaptura de la mayor parte del glutamato sináptico (79). De esta manera podría atenuarse la deficiencia de glutamato sináptico, por una permanencia mayor en la hendidura sináptica; pero se requieren más observaciones para ratificar estos hallazgos.
No en todas las regiones corticales de pacientes esquizofrénicos se presentan los mismos cambios adaptativos en el sistema glutamatérgico. Es importante señalar que en el hipocampo, estructura fundamental del sistema límbico y con extensas relaciones con la corteza prefrontal, la amígdala y el núcleo acumbens, se presentan adaptaciones moleculares compatibles con una hiperfunción glutamatérgica, por ejemplo, disminución en el enlace de receptores AMPA y kainato y disminución del mRNA para algunos receptores no NMDA. Sin embargo, no hay cambios para el transportador EAAT2.
Agradecimientos
Esta publicación es parte del proyecto Evolución temporal de los cambios en la corteza cerebral inducidos por isquemia. Los autores desean expresar su agradecimiento a la Vicedecanatura de Investigaciones de la Facultad de Salud y a la Vicerrectoría de Investigaciones de la Universidad del Valle, por su apoyo. Igualmente, a los miembros del Centro de Estudios Cerebrales, por sus comentarios y sugerencias.
Bibliografía
1. Davenport HW. Early history of the concept of chemical transmission of the nerve impulse. Physiologist 1991;34:129- 90. [ Links ]
2. Zucker RS. Exocitosis: a molecular and physiological perspective. Neuron 1996;17: 1049-55. [ Links ]
3. De Camili P, Takei. K. Molecular mechanisms in synaptic vesicle endocytosis and recycling. Neuron 1996;16:481-6. [ Links ]
4. Squire LR. Memory and the hippocampus: a synthesis from findings with rats, monkeys and humans. Psicol Rev 1992;99:195-231. [ Links ]
5. Herman JP, Culliman WE. Neurocircuitry of stress: central control of the hypothalamo- pituitary-adreno cortical axis. Trends in Neuroscience 1997;20:78-84. [ Links ]
6. Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 1999; 38:1431-76. [ Links ]
7. Fagg GE, Foster AC. Amino acid neurotransmitters and their pathways in the mammalian central nervous system. Neuroscience 1983;9:701-19. [ Links ]
8. Fonnum F. Regulation of the synthesis of the transmitter glutamate pool. Prog Biophys Mol Biol 1993;60:47-57. [ Links ]
9. Hamberger AC, Chiang GH, Nylen ES, Cotman CW, et al. Glutamate as a CNS transmitter I: evaluation of glucose and glutamine precursors for the synthesis of the prefferentially selected glutamate. Brain Research 1979; 168:513-30. [ Links ]
10. Mc Cormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Progress in Neurobiology 1992;39: 337-88. [ Links ]
11. Mesulam MM, Hersch LB, Mash DC, Geula C. Differential cholinergic innervation within functional subdivisions of the human cerebral cortex: a choline acetyltransferasa study. J Comp Neurol 1992;318:316-28. [ Links ]
12. Lewis DA, Gonzales-Burgos G. Intrinsic excitatory connections in the prefrontal cortex and the pathophysiology of schizophrenia. Brain Research Bulletin 2000;52(5):309-17. [ Links ]
13. Giorguieff MF, Glowinski ML. Presynaptic effect of L-glutamic acid on the release of dopamine in rat striatal slices. Neuroscience 1997;6:73-7. [ Links ]
14. Porras A, Sanz B, Mora F. Dopamineglutamate interactions in the prefrontal cortex of the conscious rat: studies on aging. Mech Aging Dev 1997;99:9-17. [ Links ]
15. Szethagotai. Architecture of the cerebral cortex. In: Taiper H, Word A, Pope A, editors. Basic mechanisms of epilepsies. Boston: Little Brown; 1969. p. 13-28. [ Links ]
16. Hendry SHC, Jones EG. Sizes and distribution of intrinsic neurons incorporating tritiated GABA in monkey sensory-motor cortex. J Neuroscience 1981;1:390-408. [ Links ]
17. Benes FM, Barretta S. Gabaergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001;25(1):1-27. [ Links ]
18. Goldman-Rakic P. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Society of Biol Psychiatry 1999; 46:650-61. [ Links ]
19. Benes FM. Emerging principles of altered neural circuitry in schizophrenia. Brain Res Rev 2000;31:251-69. [ Links ]
20. Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowiski J. Dopamine D1/ D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. PNAS 2001;98(1):301-6. [ Links ]
21. Carlsson A, Waters N, Carlsson ML. Neurotransmitter interactions in schizophrenia therapeutic implications. Society of Biol Psychiatry 1999;46:1388-95. [ Links ]
22. Parent A, Hazrati LN. Functional anatomy of the basal ganglia I: the cortico-basal ganglia-thalamo-cortical loop. Brain Res Rev 1995;20:91-127. [ Links ]
23. Sadikot A, Parent A, Francois C. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a PHA-L study of subcortical projections. J Comp Neurol 1992b; 315:137-59. [ Links ]
24. Paquet M, Smith Y. Differential localization of AMPA glutamate receptor subunits in the two segments of the globus palidus and the substantia nigra pars reticulata in the squirrel monkey. Eur J Neurosci 1996;9:2219-24. [ Links ]
25. Betabet R, Greenamyre JT. Differential expression of glutamate receptors by the dopaminergic neurons of the primate striatum. Exp Neurol 1999;159:401-8. [ Links ]
26. Hirsch SR, et al. A pivotal role for glutamate in the pathogenesis of schizophrenia, and its cognitive function. Pharmacology Bioch and Beba 1997;56(4):797-802. [ Links ]
27. Goff DC, Wine L. Glutamate in schizophrenia: clinical and research implications. Schiz Res 1997;27:157-168. [ Links ]
28. Escobar MI, Pimienta HJ. Áreas corticales en sistema nervioso:neuroanatomía funcional, neurohistología, neurotransmisores, receptores y clínica. Cali: Universidad del Valle; 1998. [ Links ]
29. Castro-Alamancos MA, Connors BW. Thalamocortical synapses. Prog Neurobiol 1997; 51:581-606. [ Links ]
30. Rempel-Clower N, Barbas H. The laminar pattern of connections between prefrontal and anterior temporal cortices in the Rhesus monkey is related to cortical structure and function. Cerebral Cortex 2000;10:851-65. [ Links ]
31. Marín-Padilla M. Desarrollo de la corteza cerebral humana: teoríacito arquitectónica. Rev Neurol 1999; 29(3):208-16. [ Links ]
32. Somogyi P, Tamás G, Lujan R, Jul E. Salient features of synaptic organization in the cerebral cortex. Brain Res Rev 1998;26:123-35. [ Links ]
33. Lewis DA, Gonzales-Burgos G. Proceedings of the human cerebral cortex: from gene to structure and function. Brain Res Bull 2000;52(5):309-17. [ Links ]
34. Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia- implications for schyzophrenia: an open-label study. Trends in Neurosci 1990;13:272-6. [ Links ]
35. Fitzgerald LW, Deutch AY, Gasic G, et al. Regulation of cortical and subcortical glutamate receptor subunit expression by antipsychotic drugs. J Neurosci 1995;15: 2453-61. [ Links ]
36. White B, Sullivan J, De Gracia D, O´Neil B, Neumar R, Grossman L, et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. Journal of the Neurological Sciences 2000;179:1-33. [ Links ]
37. Ramassubu R. Relationship between depression and cerebrovascular disease: conceptual issues. Journal of Affective Disorders 2000;57:1-11. [ Links ]
38. Arango C, Pimienta HJ, Escobar MI. Post stroke depression: a physiopathological an clinical approximation. Rev Col de Psiq 2000;34(4):321-44. [ Links ]
39. Chih ChP, Lipton P, Roberts E. Do active cerebral neurons really use lactate rather than glucose. Trends in Neuroscience 2001;24(10):573-8. [ Links ]
40. Seal RP, Amara SG. Excitatory amino acid transporters: a family in flux. Annual Review of Pharmacology and Toxicology 1999;39:431-56. [ Links ]
41. Gegelavishi G, Shousboe A. High affinity glutamate transporters: regulation of expresion and activity. Molecular Pharmacology 1997;52:6-15. [ Links ]
42. Palmada M, Centelles J. Excitatory aminoacid neurotransmision: pathways for metabolism, storage and reuptake of glutamate in the brain. Frontiers in Bioscience 1998;20:701-18. [ Links ]
43. Zhang Y, Kanner B. Two serine residues of the glutamate transporter GLT 1 are crucial for coupling the fluxes of sodium and the neurotransmitter. PNAS 1999;96:1710-5. [ Links ]
44. Sibson N, Dhankar A, Mason G, Rothman D, Behar K, Shulman R. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. PNAS 1998; 95:316-21. [ Links ]
45. Rauen T, Wiebner M. Fine tuning of glutamate uptake and degradation in glial cells: common transcriptional regulation of GLAST and GS. Neurochemistry International 2000;37:179-89. [ Links ]
46. Carmignoto G. Reciprocal communication between astrocytes and neurons. Progress in Neurobiology 2000;62:561- 81. [ Links ]
47. Pitts FN, McClure JN. Lactate metabolism in anxiety neurosis. New England Journal of Medicine 1967;277:1329-36. [ Links ]
48. Dager SR, Richards T, Strauss W, Artru A. Single-voxel H-MRS investigation on brain metabolic changes during lactate-induced panic. Psychiatry Research 1997;76:89- 9. [ Links ]
49. Conn J, Pin JP. Pharmacology and function of metabotropic glutamate receptors. Annu Rev Pharm and Tox 1997;37:205- 37. [ Links ]
50. Krieger P, Hellgren J, Kettunnen P, Jabbar, A. Interactions between metabotropic and ionotropic glutamate receptors regulates neuronal network activity. The Journal of Neuroscience 2000; 20(14):5382-91. [ Links ]
51. Yermolaieva O, Brot N, Weissbach H, Heinnemann S, Hoshi T. Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. PNAS 2000; 97(1):448-53. [ Links ]
52. Seeburg PH, Monyer H, Sprengel R, Burnashev N. Molecular biology of NMDA receptors. In: Collingridge GL, Watkins, JC, editors. The NMDA receptor. 2nd ed. Oxford University Press; 1994. p. 147-57. [ Links ]
53. Hansson E, Muyderman H, Leonova J, Allason L, Sinclair J, Blostrand F, et al. Astroglia and glutamate in physiology and pathology: aspects on glutamate transport, glutamate-induced cell swelling and gap-junction communication. Neurochemistry International 2000; 37:317-29. [ Links ]
54. Danbolt NC. Glutamate uptake. Progress in Neurobiology 2001;65:1-105. [ Links ]
55. Tanaka K. Functions of glutamate transporters in the brain. Neuroscience Research 2000;37:15-9. [ Links ]
56. Bruhn T, et al. Ischemia induced changes in expression of the astrocyte glutamate transporter GLT1 in hippo-campus of the rat. Neurochemistry International 2000; 37:277-85. [ Links ]
57. Martin LJ, Brambrink A, Lehmann C, Portera- Cailliau C, Rothstein J, Traysmant R, et al. Hypoxia ischemia causes abnormalities in glutamate transporters and death of astroglia in newborn striatum. Annals of Neurology 1997;42(3):335-48. [ Links ]
58. O´Regan MH, Ren J, Philis JW. Transporter reversal as a mechanism of glutamate release from the ischemic rat cerebral cortex: studies with dl threo-betabenzyloxyaspartate. Brain Research 2000;868(1):105-12. [ Links ]
59. Diemer NH, et al. Ischemia induced changes in expression of the astrocyte glutamate transporter GLT 1 in hippocampus of the rat. Neurochemistry International 2000;37(2-3):227-85. [ Links ]
60. Takanshi M, Billups B, Rossi D, Sarantis M, Hamman M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. Journal of Experimental Biology 1997;200(Pt 2):401-9. [ Links ]
61. Rossi D, Oshima T, Atwll D. Glutamate release in severe brain ischemia is mainly by reverse uptake. Nature 2000;403(6767): 316-21. [ Links ]
62. Li S, Mealing G, Morley P, Stys P. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+ dependent glutamate transport. Journal of Neuroscience 1999;19(16):1-9. [ Links ]
63. Liévens JC, Salin P, Had-Assouni L, Mahy N, Kerkerian L. Differential effects of corticostriatal and thalamocortical deafferentation on expression of the glutamate transporter GLT1 in the rat striatum. Journal of Neurochemestry 2000;74:909- 19. [ Links ]
64. Cowburn R. Neurotransmitter receptor G protein-mediated signal transduction in Alzheimer´s disease. In: Reith MEA, editor. Cerebral signal transduction: from first to fourth messengers. Totowa (NJ): Humana Press Inc; 2000. p. 129-49. [ Links ]
65. Nestler E, Hyman S, Malenka R. Memory and dementias. In: Molecular neuropharmacology: a foundation for clinical neuroscience. McGraw Hill; 2001. p. 453- 78. [ Links ]
66. Jellinger K, Stadelmann C. Problems of cell death in neurodegeneration and Azheimer´s disease. Journal of Alzheimer´s Disease 2001;3:31-40. [ Links ]
67. Frandsen A, Bisgaard J, Schousboe A. The role of second messengers in neurodegeneration. In: Reith MEA, editor. Cerebral signal transduction: from first to fourth messengers. Totowa (NJ): Humana Press Inc; 2000. p. 207-19. [ Links ]
68. Sánchez C, Diaz-Nido J, Ávila J. Phosphorylation of microtubule-associated protein 2 (MAP 2) and its relevance for the regulaton of the neuronal cytoskeleton function. Progress in Neurobiology 2000;61:133-68. [ Links ]
69. Kosik K. A notable cleavage: winding up with beta amyloid. PNAS 1999; 96:2574-6. [ Links ]
70. Saito K, Elce J, Hamos J, Nixon R. Widespread activation of calcium activated neutral proteinase (calpain) in the brain in Alzheimer´s disease: a potential molecular basis for neuronal degeneration. PNAS 1993;90:2628-32. [ Links ]
71. Whilhelsem K. The tangled biology of Tau. PNAS 1999;96:7120-1. [ Links ]
72. Alonso A, Tanweer Z, Novack M, Grunge- Iqbal I, Iqbal K. Hipephosphorylation induces self assembly of tau in to tangles of paired helical filaments/straight filaments. PNAS 2001; 28(12):6923-8. [ Links ]
73. Mattson M. Antigenic changes similar to those seen in neurofibrillary tangles are elicited by glutamate and calcium influx in cultured hippocampal neurons. Neuron 1990;4:105-8. [ Links ]
74. Van der Zee E, Douma B, Disterhoft J, Luiten P. Protein kinase C in learning and memory. In: Reith MEA, editor. Cerebral signal transduction: from first to fourth messengers. Totowa (NJ): Humana Press Inc; 2000. p. 105-25.
[ Links ] 75. Guo Q, Sopher B, Furukawa K, Pham D, Martin G, Mattson M. Alzheimer's presinilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta peptide: involvement of calcium and oxyradicals. The Journal of Neuroscience 1997; 17(11):4212-22. [ Links ] 76. Crilli M, Diodate E, Lozza C, Brussa R, Cassarini M, St George P, et al. Presinilin 1 regulates the neuronal threshold to excitotoxicity both physiologically and pathologically. PNAS 2000;97(23):128-45. [ Links ] 77. Keller J, Guo Q, Holtsberg FW, Bruce- Keller AJ, Mattson M. Increased sensitivity to mitocondrial toxin-induced apoptosis in neural cells expressing mutant presinilin- 1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci 1998;18(12):4439- 50. [ Links ] 78. Xia W, Ray W, Ostaszewsky B, Rahmati T, Kimberly W, Wolfe M, et al. Presinilin complexes with the C-terminal fragment of amyloid precursor protein at the sites of amyloid beta protein generation. PNAS 2000;97(16): 9299-9304. [ Links ] 79. Barger SW, Harmon AD. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997; 388 (6645): 878-81. [ Links ] 80. Lipton P. Ischemic cell death in brain neurons. Am Physiologycal Society 1999;70(4): 1432-1532. [ Links ] 81. Levin S, Bucci TJ, Cohen SM, Fix AS, Hardisty JF, Legrand EK, et al. The nomenclature of cell death: recommendations of an ad hoc committee of the society of toxicologic pathologists. Toxicol Pathol 1999;27:484- 90. [ Links ] 82. Rajkowska G. Postmortem studies in mood disorders indicated altered numbers of neurons and glial cells. Society of Bilologycal Psychiatry 2000;48:766-77. [ Links ] 83. Benes F. Emerging principles of altered neural circuitry in schizophrenia. Brain Research Reviews 2000;31:251-69. [ Links ] 84. Reynolds GP, Zhang ZS, Beasley CL. Neurochemical correlates of cortical gabaergic deficits in schizophrenia: selective losses of calcium binding proteins inmunoreactivity. Brain Research Bulletin 2001;55(5):579-84. [ Links ] 85. Kostovic I, Rakic P. Cytology and time of origin of interstitial neurons in the white matter in infant and adult human and monkey telencephalon. J Neurocytology 1980;9: 219-42. [ Links ] 86. Mednick SA, Machon RA, Huttunen MD. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psichiatry 1988;45:189-92. [ Links ] 87. Ingvar DH, Franzen G. Distribution of cerebral activity in chronic schizophrenia. Lancet 1974;2:1484-6. [ Links ] 88. Ebmeier KP, Lawrie SM, Blackwood DH, Johnstone, Goodwin GM. Hypofrontality revisited: a high resolution single photon emission computed tomography study in schizophrenia. J Neurol Neurosurg Psychiatry 1995;58:452-6. [ Links ] 89. Lewis DA, Gonzales-Burgos, G. Proceedings of the human cerebral cortex: from gene to structure and function. Brain Research Bulletin 2000;52(5):309-17. [ Links ] 90. Andreasen NC, Flashmau L, Flaun M, Arndt S, Swayze V, O´Leary DS, et al. Regional brain abnormalities in schizophrenia measured with magnetic resonance imaging. JAMA 1994; 272:1763-9. [ Links ] 91. Breier A, Buchannan RW, Elkashef A, Munson RC, Kirkpatrick B, Gellad F. Brain morphology and schizophrenia: a magnetic resonance imaging of limbic prefrontal cortex and caudate structures. Arch Gen Psychiatry 1992;49:921-6. [ Links ] 92. Shelton RC, Karson CN, Dovan AR, Pickar D, Bigelow LB, Weinberger DR. Cerebral structural pathology in schizophrenia: evidence for a selective prefrontal cortical defect. Am J Psychiatry 1988;145:154-63. [ Links ] 93. Lewis DA, Pierry JN, Volk DW, Melchitzky DS, Tsung-Ung W. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Society of Biological Psychiatry 1999;46:616-26. [ Links ] 94. Benes F, Berretta S. Gabaergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsichopharmacology 2001;25(1):1-27. [ Links ] 95. Ohnuma T, Auguod SJ, Arai H, McKenna PJ, Emson PC. Expression of the excitatory aminoacid transporter 2 and metabotropic receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Mol Brain Res 1998;56:207-17. [ Links ] 96. Arriza WA, Fairman JJ, Wadiche GH, Murdoch MP, Kavanaugh SG. Functional comparisons of the three glutamate transporters subtypes cloned from human motor cortex. J Neurosc 1994; 14:5559- 69. [ Links ] 97. Kornhuber J, Mack-Burkhardt F, Riederer P, Hebenstreit GF, Reynolds GP, Andrews HB, et al. 3[H] MK-801 binding sites in postmortem brain regions in schizophrenia patients. J Neural Transm 1989;77:231-6. [ Links ] 98. Nishikawa T, Takashima M, Toru M. Increased 3[H] rainic acid binding in the prefrontal cortex in schizophrenia. Neuroscience Letters 1983;40:245-50. [ Links ] 99. Simpson MD, Slater P, Royston MC, Deakin JF. Alterations in phenylciclidine and sigma binding sites in schizophrenia brains. Effect of disease process and neuroleptic medication. Schizoph Res 1991;6:41-8. [ Links ]

