










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.32 no.1 Bogotá Jan./Mar. 2003
Epistemología, filosofìa de la mente y bioètica
Dilemas éticos sobre el uso de placebo en investigación terapéutica
Álvaro Franco, M.D.
Psiquiatra infantil y de adolescentes, magíster en Bioética y director de la División de Investigaciones, Universidad El Bosque, Bogotá.Resumen
El presente artículo presenta una reflexión desde la bioética acerca del uso de las llamadas sustancias placebos en los ensayos clínicos terapéuticos, realizados en sujetos humanos. Se parte de una definición del concepto placebo; luego se presenta una rápida revisión de los aspectos esenciales de la investigación terapéutica y de los conflictos éticos generados, pero se hace hincapié en la investigación cooperativa internacional y en sus efectos en los países en desarrollo; por último, se concluye que el consentimiento informado como elemento significativo del respeto a la dignidad humana se convierte en la piedra angular para el uso ético de placebos, y corresponde a los comités éticos de investigación intervenir activamente, tanto en el consentimiento informado como en la regulación de su uso.
Palabras clave
Placebo, bioética, investigación terapéutica.
Abstract
The purpose of this work is to show the most relevant aspects of a research the ethical aspects of the use of placebo, both in the clinical and the research fields, and concludes that consent information from the patient is the most important aspect the ethical use of placebo and evaluation from Ethics committees the use of placebo and the research ethics committees has to participate actively and regular for use of placebos.
Key words
Placebo, bioethics, therapeutical research.
Introducción
Etimológicamente la palabra placebo procede del latín placebo, que significa complacer. Para Pedro Laín Entralgo (1) la primera definición con un sentido médico se encuentra en el Quincy Lexicon, de 1787, complementada en el Hooper Medical Dictionnary, de 1811, y definida así: «medicación prescrita para complacer al enfermo». En el común de las personas placebo es una sustancia inerte que se les da a los pacientes «que son molestos, exigentes o a quienes no hay nada que hacerles», pero en realidad este concepto ha variado, y sólo hasta 1950 A. D. Berg, al separar placebo de su efecto, afirma: «toda intervención terapéutica, incluido el propio placebo tiene efecto placebo, siendo además esta la única característica común a todos los medicamentos» (2). Para el presente trabajo acojo la definición del Consejo de Investigaciones del Canadá: «Algún procedimiento o componente terapéutico que es dado intencionadamente buscando tener un efecto, en un paciente, síntoma, síndrome, o enfermedad, pero objetivamente sin actividad específica para esa condición tratada» (3).
En el ámbito de la investigación biomédica el uso de placebo está restringido al sometimiento dado para probar un nuevo fármaco o procedimiento terapéutico con el fin de determinar su eficacia, esto es, si sirve o no para una determinada indicación o, en caso de los medicamentos, si es mejor o no con nuevas vías de administración (oral, parenteral, transcutánea, etc.). Después se comparan los grupos experimentales y los de control con los resultados obtenidos por este último con un placebo.
Los individuos que sean sujetos de investigación deben cumplir con determinados criterios de inclusión; luego de esto deben de ser explicados el procedimiento, los riesgos que conlleva y los objetivos de la investigación, para que dichos individuos puedan tomar la opción de hacer parte del grupo de investigación o no, lo que se ve expresado en el consentimiento informado. En este punto los investigadores deben asignar al azar los grupos experimentales y los de control, a fin de realizar el estudio en condiciones ciegas, que pueden ser tanto ciego simple, cuando el sujeto no conoce en qué grupo quedó, si en el que recibirá placebo (control) o en el grupo que tomará el tratamiento en investigación (experimental), como doble ciego, cuando ni el sujeto ni el investigador conocen el grupo en el cual fueron asignados los participantes. Los datos obtenidos deben someterse a procedimientos estadísticos validados, en los que se acepta como norma general una diferencia observada mayor del 5% como significativo, esto es, la probabilidad de que el azar explique la diferencia es menor del 0,05, por lo que es más significativo en la medida en que la muestra sea más grande. También la diferencia debe ser clínicamente relevante.
Dilemas éticos comunes en la investigación con placebo
Existen varios dilemas en torno al uso de placebos en investigación biomédica, pero para De Abajo y Gracia el problema ético fundamental se plantea así: «¿Se puede someter a un hombre a riesgos notables, mortales quizá, con el simple propósito de aumentar nuestro conocimiento sobre algo?».(2). Citan los mismos autores que históricamente ha evolucionado la respuesta a este interrogante: en Alejandría, en el siglo I (d. C.) se utilizaban los condenados a muerte para fines experimentales; Bernard sólo justificaba la experimentación en animales y en cadáveres, y sir William Osler la defiende siempre y cuando acepten voluntariamente los sujetos una vez sean informados de los riesgos que puedan correr. La respuesta al interrogante se cifra en el cumplimiento del canon o patrón que nos permite juzgar racionalmente lo ético de la conducta o, en este caso, de la investigación:
- Principio de reciprocidad: o regla de oro, en la cual el mandato es no hacer a los demás, lo que no queramos que éstos hagan con nosotros.
- Principio de universalidad: si una acción concreta es correcta o incorrecta, tiene que afirmar a la vez que todas las acciones exactamente iguales a la juzgada deben tomarse también como correctas o incorrectas.
Hoy el punto de referencia puede ser, de acuerdo con mi criterio, el Informe Belmont (4) y la publicación de la primera edición del libro Principios de ética biomédica (5), de Beauchamp y Childress, convertido ya en un clásico contemporáneo del cual abordaré algunos dilemas que se presentan tanto en la investigación farmacológica como en la psicoterapéutica.
Calidad de los investigadores
El investigador responsable o grupo de investigadores, en caso de laboratorios farmacéuticos, deben acreditar su competencia y solvencia en el campo, y para esto existen varios parámetros, entre los que podemos citar:
- Formación académica.
- Trayectoria como investigador (es). - Publicaciones en revistas indexadas.
- Explicitación de los compromisos comerciales.
- Reconocimiento de los beneficios
- que ha(n) obtenido por las casas farmacéuticas.
Selección equitativa de las muestras
Las muestras deben exponer paridad en los grupos seleccionados, tanto en los escogidos por la patología como en los controles. Los primeros deben tener similar estado clínico en su evolución, factores de pronóstico, gravedad e intensidad de la sintomatología, periodo de evolución, etc. En las características de los sujetos objeto de la investigación no debe existir discriminación alguna (color, edad, sexo, religión, etc.) y la asignación debe ser completamente al azar.
Información veraz a los sujetos de investigación
La información veraz implica que los sujetos de investigación que han accedido voluntariamente al proyecto tienen una comprensión total de los riesgos, de los beneficios y de los objetivos de la investigación; conocen la posibilidad de quedar al azar en el grupo experimental o en el grupo de control (placebo); aceptan participar luego de hacer las preguntas que consideren necesarias, y, además, están seguros de que pueden retirarse en cualquier momento, sin ningún tipo de represalias, y de que los riesgos están totalmente cubiertos por el grupo investigador responsable.
Doble papel de médico e investigador
Como investigador, el médico actúa para generar conocimiento científico previsto para beneficiar a futuros pacientes. Como clínico en ejercicio, los contratos y la responsabilidad de atender requieren actuar por el mejor interés de las pacientes actuales. Ambos papeles pretenden beneficiar a los enfermos. Según esto, las responsabilidades con las futuras generaciones pueden entrar en conflicto con el cuidado debido a los pacientes de hoy. Dilemas éticos
La investigación en seres humanos es tan importante como moralmente peligrosa para la sociedad, porque los sujetos están expuestos a algún riesgo para promover el avance de la ciencia. La investigación éticamente justificada debe satisfacer varias condiciones, aun las que incluyen la búsqueda de conocimiento, una expectativa razonable de que la investigación generará el conocimiento que se busca, un balance favorable de beneficios para el sujeto y para la sociedad sobre los riesgos a los que el sujeto se expone, una selección equitativa de los sujetos y la necesidad de emplear seres humanos. El consentimiento es necesario, pero por sí solo no es suficiente para justificar la investigación.
Autores como De Deyn y D’Hooge (6) consideran que el control con placebo se puede incluso considerar una obligación ética, pero el médico que participa en investigación debe tener presente algunas cláusulas. Éstas son:
-No debe existir ninguna terapia adecuada para la enfermedad ni presumirse que existan efectos secundarios serios.
- El tratamiento con placebo no debe durar demasiado tiempo.
- El tratamiento con placebo no debe infligir riesgos inaceptables.
- El sujeto de experimentación debe ser adecuadamente informado y dar su consentimiento.
Otro conflicto se presenta cuando el médico, en su doble papel de médico e investigador, en estudios doble ciego, no sabe si un paciente está tomando la droga o el placebo y ante síntoma de aparición reciente no se puede saber con exactitud si el paciente está sufriendo un efecto secundario de la medicación o está frente a un nuevo problema médico (7).
Conflictos en los ensayos clínicos
A veces se necesitan los ensayos clínicos controlados para confirmar que un efecto observado, como una mortalidad reducida para una enfermedad determinada, es consecuencia de una intervención concreta, más que el resultado de una variable desconocida en la población de pacientes. Si existen dudas acerca de la eficacia o de la seguridad de un tratamiento o sobre sus relativos méritos en comparación con otro tratamiento, está claramente justificada la investigación científica que las pueda resolver. Los ensayos controlados son instrumentos científicos que pretenden proteger a los pacientes actuales y futuros del entusiasmo y las corazonadas médicas, al sustituirlas por tratamientos inequívocos de validez comprobada. Enmascarar información al médico-investigador, de acuerdo con algunas interpretaciones, también desempeña una función ética, porque obvia cualquier conflicto de intereses para aquellos que a la vez brindan terapia y realizan investigación. En el caso específico de los ensayos clínicos aleatorios (ECA), no existe justificación para dejar de revelar a los potenciales sujetos todas las series de métodos, tratamientos y placebos que su usarán; sus riesgos y beneficios conocidos, y cualquier incertidumbre conocida. La investigación no estará justificada si hay duda significativa sobre la materialidad de la información no revelada o si están disponibles diseños de investigación alternativos científicamente satisfactorios.
Para Evans (8), la utilización de placebos en los ECA incluye la posibilidad de engaño, pero su utilización en investigaciones clínicas se justifica si cumplen las siguientes condiciones:
- Mantener la incertidumbre que se genera al distribuir aleatoriamente a los participantes en diferentes regímenes de tratamiento.
- Aclarar, por parte de los investigadores, los mecanismos de distribución aleatoria de los pacientes que participaron en el estudio, así como las probabilidades que tuvieron para estar recibiendo placebo o no.
El mismo autor, basado en las consideraciones anteriores, concluye que los estudios de simple ciego, donde el paciente pasa por periodos en que toma el principio activo y otros en los que no recibe tratamiento, no se cumplen los criterios anteriores, por lo tanto, esta práctica parece no ser justificable desde el punto de vista ético.
Condiciones de justificación de los ensayos clínicos aleatorizados
El compromiso del científicoinvestigador es eliminar terapias inferiores y peligrosas y ofrecer las que han sido contrastadas y han resultado más beneficiosas, tanto para los pacientes actuales como para los futuros. Los riesgos para algunos están justificados por los beneficios claramente mayores que se anticipan para otros. Beauchamp y Childress (5) consideran que los ECA lo están sólo en caso de cumplir las siguientes condiciones:
-El ensayo se diseña como un experimento crucial entre alternativas terapéuticas y encierra una promesa científica de conseguir un resultado.
- Un comité ético de investigaciones aprueba el protocolo y certifica que ningún médico tiene conflicto alguno de interés ni incentivo que pudiera amenazar la relación médico-paciente.
- Se obtiene un consentimiento informado exhaustivo
- No puede usarse placebo si existen pruebas de que hay un tratamiento adecuado.
- Un comité de seguimiento de datos pondrá fin al ensayo cuando el equilibrio clínico se desplace por datos estadísticamente significativos o, bien, suministrará a los médicos y a los pacientes información significativa, suficiente para que una persona razonable decida permanecer en el ensayo o retirase de él.
- Se protege en todo momento el derecho de los médicos de recomendar la retirada y el derecho de los pacientes a retirarse.
La investigación con placebos en países en desarrollo
De acuerdo con el principio de universalidad ya comentado, los estándares éticos no pueden cambiar de un país a otro. En el llamado tercer mundo existen ‘facilidades’ para que protocolos no autorizados por razones éticas en otras latitudes sí se puedan realizar en nuestros países.
En Latinoamérica, un estudio realizado por el Programa Regional de Bioética OPS-OMS, citado por Manzini (9), mostró una ausencia total de normativas y de comités de ética en diez países de la región (48% de la muestra), mientras que en seis (29%) no se cumple el requisito de la evaluación de protocolos de investigación.
Otros aspectos relacionados son los citados en el documento Estándares éticos y científicos en la investigación, editado por la Fundación Grifols de Barcelona (10), los cuales están relacionados, por un lado, con factores generales, como el analfabetismo, la pobreza, el creciente número de enfermedades (en especial infecciosas dada la baja cobertura de saneamiento ambiental), etc. y, por otro, con algunos factores específicos, como lo son las motivaciones de los patrocinadores con relación a las expectativas de los investigadores, la necesidad de evitar la explotación mediante la propiedad selectiva de los datos y de su uso (lo cual incluye el problema de la propiedad intelectual y la necesidad de garantizar que no se usaran individuos vulnerables) e impedir que en un futuro los pacientes no tengan acceso a los beneficios que puedan reportar las investigaciones.
Además de los anteriores, hay otros dilemas que se presentan en investigaciones originales, producto de los países en desarrollo, por ejemplo, que el investigador piense que está suministrando un medicamento activo, cuando en realidad proporciona una sustancia sin el efecto terapéutico buscado, pero con el nombre genérico de lo que cree estar utilizando, dada la mala calidad y la ausencia de estudios de biodisponibilidad y de bioequivalencia en algunas copias de productos originales, es decir, sin saberlo podría estar enfrentando placebo contra placebo.
Los problemas éticos más comunes que he observado en la revisión de protocolos colombianos con medicaciones psiquiátricas son los siguientes:
- Enfrentar medicamentos de eficacia ya establecida contra placebo, lo que no tiene ninguna justificación ética.
- Medir su la validez sin compararlo (aleatoriamente) con un producto similar, con el fin de determinar cuál es el superior.
- Estudiar la eficacia en la población colombiana sin tener en cuenta los mínimos aspectos etnofarmacológicos en el protocolo, como el de incluir la etnia: mestizo (la mayoría de nosotros), blanco, negro, indio, zambo, etc.
- Plantear que «El estudio ha sido diseñado siguiendo estrictamente los criterios éticos de la Declaración de Helsinki», pues se ha comentado que seguir de manera ‘estricta’ estos criterios no da margen para las investigaciones novedosas.
- No figurar el aval de ningún comité ético de investigación. ! Pasar por alto problemas en el diseño metodológico.
- Cobrar por los procedimientos del estudio, por ejemplo, establecer que «Cada paciente recibirá las primeras tabletas de...», pues en éstos se busca hacer mercadeo; además de que no se plantea una mínima compensación a los pacientes que participan en el estudio, como lo es suministrarle el medicamento completamente gratis hasta cuando el tratante lo considere necesario. Honorarios: Aún cuando es un punto de discusión, la mayor parte de laboratorios internacionales compensan la tarea del médico con tarifas variables por paciente incluido dentro del estudio; estas tarifas se han vuelto una costumbre, pues evitan la compensación en especie. Con el nombre de Investigación Colaborativa Internacional, las multinacionales farmacéuticas han incursionado cada vez con más frecuencia en nuestros países para completar las fases del desarrollo de sus nuevos productos, Mainetti se pregunta «¿Quiénes deben recibir los beneficios de la investigación y quiénes sus perjuicios?» (11). La contrapregunta se da en los términos siguientes: ¿los países pobres deberían renunciar a los beneficios económicos que conllevan estas investigaciones? En mi concepto personal, tendríamos que llegar a una situación de equilibrio donde los comités éticos de investigación locales fijen en cada caso particular la recomendación de aceptación o no de un protocolo. La Comisión Nacional de Bioética de los Estados Unidos ha publicado un documento (12) en el que, entre otras cosas, prohíbe a las compañías farmacéuticas que prueben nuevos medicamentos en comunidades pobres sin que sus residentes se beneficien de los medicamentos si se descubre que éstos son seguros y eficaces. El informe hace las siguientes recomendaciones:
- Los protocolos de investigación tienen que ser aprobados en los EE.UU. y también en el país donde se vayan a realizar los estudios.
- Los investigadores estadounidenses que trabajen en países del tercer mundo no deben utilizar placebos cuando hay productos activos eficaces para la enfermedad cuya cura se busca.
- Los investigadores tienen que consultar con los miembros de la comunidad donde se va a llevar a cabo el estudio, desde que se inicia su diseño, para asegurar que se toman en cuenta las necesidades de la comunidad.
- Los investigadores tienen que obtener confirmación de que los que participen en la investigación han aceptado ser parte del estudio.
- Como se ha analizado previamente, la aprobación sobre el desarrollo o no de un estudio la Franco, A., M.D.
deben dar los comités de ética locales.
Comités éticos de investigación (CEI) Con el fin de garantizar una consciente y libre participación de los voluntarios o de los pacientes en un ensayo clínico se requiere que el protocolo sea aprobado por un comité ético de investigación (CEI), el cual va verificar que éste cumple con las siguientes condiciones:
- Los riesgos deben minimizarse.
- Éstos deben ser razonables, en función de los beneficios que se esperan y la importancia de los conocimientos que puedan esperarse.
- La selección de los sujetos ha de ser equitativa.
- Se pedirá el consentimiento a cada persona en términos claramente comprensibles.
-Éste se documentará debidamente.
- El proyecto de investigación ha de incluir el seguimiento y control para garantizar la seguridad de los sujetos y el secreto profesional. Existen, además, otras recomendaciones que usualmente tienen en cuenta los CEI, como las normas de buena práctica clínica —BPC— (13). Éstas son una serie de medidas de carácter administrativo que deben ser observadas para que el informe de un ensayo clínico sea aceptado por las autoridades sanitarias (específicamente por la Food and Drug Administration), como demostración sustancial de la seguridad y eficacia de un nuevo medicamento (14).
La industria farmacéutica es la primera interesada en salvaguardar la seguridad, la eficacia y la aceptabilidad de los fármacos por las autoridades sanitarias. De aquí que la observancia de estas normas, muy detalladas, permite garantizar, desde la vertiente más objetiva y razonablemente controlable, que el ensayo es éticamente correcto y que si el comité es competente y cumple su función, es ético o de lo contrario desaparece.
En Colombia la prioridad está dada por la protección del sujeto de investigación, determinada por el riesgo —Res. 8430 de 1993 (15)—, además, por Ley 23 de 1981 (16) se deben cumplir las normas de la Declaración de Helsinki (17), promulgadas en Edimburgo, en el 2000. También se debe tener presente en la valoración ética del protocolo el consentimiento inforDilemas éticos mado. Las investigaciones con placebo en nuestro medio se deben limitar a evaluar la eficacia de un medicamento o de un procedimiento terapéutico en estudios clínicos aleatorios.
Consentimiento informado Como lo hemos visto a lo largo de la exposición, en algunas ocasiones es adecuado el uso de placebos, bajo la premisa fundamental de que quien los tome o reciba su efecto tenga total información que le permita reflexionar y optar libremente.
El consentimiento informado es la materialización del respeto a la dignidad humana y no es un simple documento, es más un proceso de comunicación bidireccional en la relación médico-paciente. Conviene entenderlo más como una consejería, en la que se deben tener en cuenta influencias como los sentimientos, las emociones, las conductas, las creencias y los conocimientos de los pacientes, para que así se permita la libre opción. De esta manera se convierte en la imagen jurídica que representa la autonomía del individuo.
La práctica y la difusión del consentimiento informado en medicina aparece en un determinado contexto sociopolítico, en el que la lucha y la defensa de las libertades individuales y la igualdad se extiende a múltiples áreas sociales (política, trabajo y educación). En la práctica asistencial disminuye la dependencia del individuo del sistema de salud y deja de ser progresivamente un sujeto pasivo sobre el que se realizan acciones terapéuticas ‘por su bien’, para convertirse en la parte interesada y decisiva de todo acto terapéutico. El consentimiento informado cobra así el carácter de derecho fundamental que condiciona toda la terapéutica, con lo que prima la autonomía del individuo sobre el antiguo paternalismo. Se ha difundido su utilización no sólo para la experimentación, sino en la aplicación para realizar pruebas diagnósticas y aceptar tratamientos. Todo médico o investigador debe obtener el consentimiento informado de sus pacientes. Esta expresión surgió diez años después de los juicios de Nuremberg, pero hasta 1972 se estudió. Su interés está centrado no sólo en la obligación del investigador o del profesional de exponer una información, sino en la calidad de la comprensión y del consentimiento del paciente o sujeto.
Broekman (18) lo define como «Derecho a obtener información en relación con todos los hechos médicamente relevantes, a ser informado y a responder a dicha información mediante el consentimiento ». Aparte de lo anterior, se considera como la piedra angular de la expresión jurídica en la relación médico-usuario (paciente con derechos). Beauchamp y Childress (5) consideran que el consentimiento informado debería ser sinónimo de una toma de decisiones conjunta, esto es un proceso temporal, no es sólo un documento firmado. Para ellos conlleva un doble significado.
Es una autorización autónoma para una determinada intervención o para participar en un proyecto de investigación. En aquellas instituciones donde es necesario obtener un consentimiento legalmente válido de parte de los pacientes constituye una autorización institucional o legalmente efectiva.
El manual de ética de la Asociación Estadounidense de Médicos define el consentimiento informado como: «La explicación, a un paciente atento y mentalmente competente, de la naturaleza de su enfermedad, así como del balance entre los efectos de la misma y los riesgos y beneficios de los procedimientos terapéuticos recomendados, para a continuación solicitarle su aprobación para ser sometido a esos procedimientos. La presentación de la información al paciente debe ser comprensible y no sesgada; la colaboración del paciente debe ser conseguida sin coerción, el médico no debe sacar partido de su potencial dominancia psicológica sobre el paciente.» (19) Con la aplicación del consentimiento informado y la garantía que pueda brindar un comité ético asistencial se puede limitar el abuso de placebos, generar grandes ahorros al sistema de salud y evitar malos tratos y sufrimientos a los pacientes.
Propuesta metodológica para el análisis de casos Para un análisis de protocolos con uso de placebo, se propone el siguiente árbol de decisiones (Grafico1).
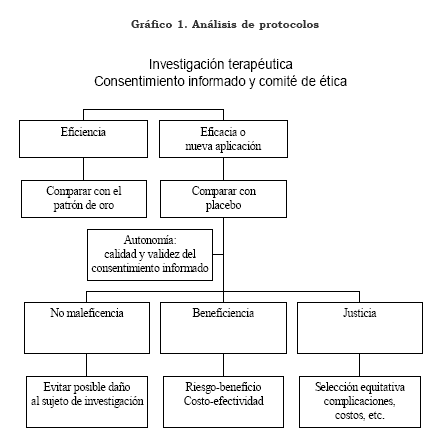
-¿Está justificada la utilización de placebos?
- Identificar los principios implicados en cada conflicto: autonomía (calidad y validez del consentimiento informado); beneficencia (¿qué utilidad busca esta investigación?, ¿para quién son los beneficios?, y ¿quién corre los riesgos? Relaciones costo- beneficio y costo-efectividad), no maleficencia (¿compromete de alguna manera la salud o el bienestar del sujeto —por ejemplo, no es adecuado utilizar un placebo en infecciones bacterianas para probar un nuevo antibiótico—?) y justicia (¿se utilizaron fondos propios, del patrocinador o del sistema de salud?).
- Determinar las características especiales de algunos pacientes: autonomía limitada (al menos temporal) y dificultades sociales, emocionales, educativas, etc. (¿participan en la investigación como opción para tener tratamiento?).
Establecer probables cursos de acción y consecuencias: ¿qué pasaría si lo recibe y qué si no lo recibe, tanto con el paciente como con la investigación (identificación del curso óptimo)? Conclusiones Desde la bioética clínica se debe reflexionar y examinar las limitaciones, los beneficios o los daños que pueda ocasionar el uso de placebo de manera indiscriminada, inadecuada o no pertinente, a fin de asegurar que su uso sea juicioso y racional por parte del médico o del investigador. Ante la necesidad de utilizar placebo para impedir conclusiones falsas sobre la eficacia de un fármaco, de un procedimiento diagnóstico o terapéutico y las consecuencias que sobre la salud pública y la justicia sanitaria pueda tener el concluir que un medicamento o que una práctica instituida por la tradición sea eficaz o no, es necesario tener procedimientos de validación adelantados con metodologías apropiadas.
Introducir un placebo y reconocer la superioridad de un tratamiento sobre éste es esencial para un estudio farmacológico quepretenda ser válido en el campo de medicina, pues usar la ausencia de tratamiento como un control no lo es. La FDA requiere demostrar la superioridad del nuevo medicamento sobre el placebo. Asimismo, desempeñan papeles fundamentales tanto el consentimiento informado en el ámbito individual como las decisiones por consenso en los comités de ética para preservar los derechos y el mejor interés de los pacientes en el colectivo.
Particularmente los CEI deben trabajar de una manera prudente y con conocimiento de la metodología científica y de los progresos en el área del protocolo que se va a evaluar, pues no se deben autorizar aquellos con uso de placebos en entidades en las cuales existen tratamientos eficaces y donde esté en juego la calidad de vida o la vida misma del paciente; el ejemplo más corriente sería en el ámbito de las enfermedades infecciosas bacterianas, si se utiliza el nuevo antibiótico, éste deberá ser confrontado con el mejor existente para el germen aislado y en ningún caso frente a placebo.
Bibliografía
1. Lain Entralgo P. Hacia una terapéutica general antropológica. En: Historia universal de la medicina. Barcelona: Masson; 1998. [ Links ]
2. De Abajo F, Gracia D. Ética del uso de placebo en investigación clínica. Investigación y Ciencia 1997 nov;90. [ Links ]
3. Lapierre Y. Ethics and placebo. J Psiq. Neurosci 1998;23(1):9-11. [ Links ]
4. Comisión Nacional del Congreso de los EE. UU. para la Protección de las Personas Objeto de la Experimentación Biomédica y de Conducta. Informe Belmont. Washington; 1978. [ Links ]
5. Beauchamp T, Childress J. Principios de ética biomédica. Barcelona: Masson; 1999. [ Links ]
6. De Deyn P, D'Hooge R. Placebos. Clin Practice and Research J of Medical Ethics 1996;22(3):140-6. [ Links ]
7. Groudine S, Lumb PD. At the coalface- medical ethics in practice: first, do no harm. Journal of Medical Ethics 1997; 23(6): 377-8. [ Links ]
8. Evans M. Justified Deception?: the single blind placebo in drug research. Journal of Medical Ethics 2000;26:188-93. [ Links ]
9. Mancini R. Normas éticas para la investigación clínica. Available from: http://www.uchile.cl/bioetica/doc/normas.htm [ Links ]
10. Fundación Grifols. Estándares éticos y científicos en la investigación. Barcelona; 1999. p. 35-6. [ Links ]
11. Mainetti JA. Bioética de la experimentación humana: experiencia de la Escuela Latinoamericana de Bioética. En: Programa Regional de Bioética OPS-OMS. Investigación en sujetos humanos: experiencia internacional, Santiago de Chile: Programa; 1999. [ Links ]
12. Comisión Nacional de Bioética, USA. Un panel de los EE. UU. sugiere cómo regular los ensayos clínicos pagados por compañías americanas en el extranjero Flaherty P. Washington Post 2001 apr 30. [ Links ]
13. Baluja I.: Bioética en ensayos clínicos. Su aplicación actual, Rev. Cubana Med Gen Integr 1988:14(4):340-6. [ Links ]
14. Food aand Drug Administration. Available from: http://www.fda.gov [ Links ]
15. República de Colombia, Ministerio de Salud, Resolución 8430 de 1993. Bogotá. [ Links ]
16. Ley 23 de 1981. Bogotá. [ Links ]
17. WMA. Declaración de Helsinki, 5th ed. Edimburgo; 2000. [ Links ]
18. Broekman J. Bioética con rasgos jurídicos. Madrid: Diles; 137-48. [ Links ]
19. American Medical Association. Manual de ética. Citado en Simón P. El consentimiento informado: historia, teoría y práctica. Madrid: Tricastela; 2000. [ Links ]

