










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.36 suppl.1 Bogotá Oct. 2007
Ángela María Iragorri Cucalón1
1 Médica neuróloga de la Clínica La Inmaculada, Clínica de Trastornos Cognitivos y de Memoria, Bogotá, Colombia.airagorri@gmail.com
Resumen
Introducción: el 20% de los pacientes que mueren con demencia antes de los 70 años de edad tienen demencia frontotemporal, un síndrome amplio con tres grandes variantes clínicas: variante frontal, demencia semántica y afasia primaria progresiva. Objetivo: presentar las características demográfi cas, la genética, el diagnóstico, la patología, el tratamiento y otras variantes de la demencia frontotemporal. Método: revisión de la literatura médica existente sobre el tema. Conclusión: la mayoría de las formas familiares de demencia frontotemporal tienen una herencia autosómica dominante y se asocian con mutaciones en el gen que codifi ca la proteína tau. Los síntomas iniciales están relacionados con cambios de personalidad, alteraciones comportamentales, del afecto, el lenguaje o las funciones ejecutivas, y se requieren imágenes cerebrales y pruebas neuropsicológicas para hacer un diagnóstico acertado. No existe en la actualidad tratamiento específi co para la demencia frontotemporal, y este se basa en el control de síntomas.
Palabras clave: demencia, inhibidores de recaptación de serotonina, proteínas tau.
Abstract
Introduction: Frontotemporal dementia constitutes a signifi cant percentage of the degenerative dementias, making up for 20% of patients who die with dementia before the age of 70. It is an extensive syndrome with three clinical variants: frontal variant, semantic dementia and primary progressive aphasia. Objective: To describe the demographic characteristics, genetics, diagnosis, pathology and treatment, as well as other types of frontotemporal dementia. Method: Review of the medical literature. Conclusions: Patients with this syndrome present with behavioral and affective symptoms, language diffi culties or executive dysfunction. An imaging study of the brain should be performed, as well as neuropsychological assessment. At present, no specifi c pharmacologic therapies have been approved for use in frontotemporal dementia.
Keywords: Dementia, serotonin uptake inhibitors, tau proteins.
Introducción
La demencia frontotemporal, aunque es menos frecuente que la enfermedad de Alzheimer, es responsable de un porcentaje importante de las demencias degenerativas: corresponde a 5%-7% de las series de autopsias y a 20% de los pacientes que mueren con demencia antes de los 70 años de edad (1-2). Sus manifestaciones varían de acuerdo con la distribución de la atrofi a en los lóbulos frontal y temporal
Arnold Pick fue quien describió inicialmente las características clínicas de un paciente con atrofi a lobar, circunscrita a los lóbulos frontal y temporal, asociada con afasia progresiva, en 1892 (1). En 1982, Mesulam propuso la expresión afasia primaria progresiva, al reportar seis casos de pacientes que presentaban una afectación aislada del lenguaje (3). Desde la descripción de los seis casos de Mesulam, cerca de una centena más se han descrito en la literatura médica (3-4). Por otra parte, Snowden y cols. (5) describieron pacientes con un daño progresivo de la memoria semántica, y propusieron la expresión demencia semántica. Luego, la literatura médica ha seguido reportando numerosos casos de demencia semántica (6-7).
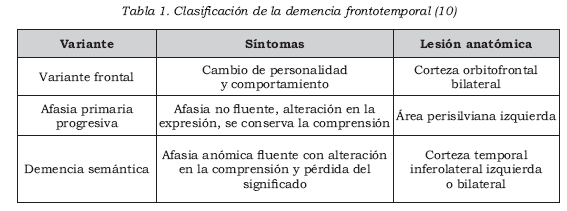
En años recientes se han logrado importantes avances en la identifi cación de características clínicas, bioquímicas, patológicas y genéticas de este grupo de enfermedades (1). Ante esta gran variabilidad de manifestaciones y luego de múltiples intentos por clasifi car la demencia frontotemporal, se ha propuesto hacerlo según su presentación clínica. Se considera un síndrome amplio con tres grandes variantes clínicas (Tabla 1): variante frontal, demencia semántica y afasia primaria progresiva (6,8-10).
Características demográficas
La demencia frontotemporal se instaura, por lo general, durante la sexta década de la vida, y la edad de muerte es usualmente en la séptima década, aunque hay reportes de casos familiares en los cuales las primeras manifestaciones de la enfermedad se presentan durante la segunda década (11). Las características demográficas difi eren de acuerdo con la variante clínica de la enfermedad:
• La variante frontal y la demencia semántica son más frecuentes en hombres, con una relación 2:1; mientras la afasia primaria progresiva es más frecuente en mujeres (11).
• La progresión de la enfermedad es más rápida en la variante frontal; la muerte se presenta a los 3,4 años en promedio. Entre tanto, los pacientes con demencia semántica viven más de seis años desde el momento del diagnóstico (12), y los pacientes con afasia primaria progresiva tienen una supervivencia intermedia. La presencia de enfermedad de motoneurona o síntomas parkinsonianos acortan el tiempo de sobrevida.
Genética
En una minoría de los casos (<10%) hay una historia familiar. La mayoría de las formas familiares de demencia frontotemporal tienen una herencia autosómica dominante y se asocian con mutaciones en el gen que codifi ca la proteína tau (relacionada con microtúbulos), localizado en el cromosoma 17 (13). En este gen se han identifi cado más de 35 mutaciones (14-16), y tres de ellas son responsables de, por lo menos, la mitad de los casos familiares de demencia frontotemporal: P301L, mutaciones del exón 10 y N279K.
La primera mutación se asocia con el fenotipo clásico de la demencia frontotemporal; la segunda, con una afectación de la memoria o el lenguaje, relacionada con parkinsonismo, y la tercera, con una degeneración palidopontonigral, que se manifi esta con parkinsonismo y parálisis supranuclear progresiva (1). Las manifestaciones clínicas encontradas con otras mutaciones incluyen ideas delirantes paranoides, comportamiento antisocial, convulsiones y, ocasionalmente, amiotrofía.
Se han descrito algunos casos de demencia frontotemporal con herencia autosómica dominante asociados con mutaciones en los cromosomas 9 (17) y 3 (18). Las mutaciones de la presenilina 1 pueden terminar en demencia frontotemporal, en ausencia de depósitos de amiloide (19). La asociación con apolipoproteína E4 es muy débil, a diferencia de lo que ocurre en la enfermedad de Alzheimer (20-21).
Diagnóstico
Los criterios diagnósticos —publicados por Neary y cols. (22)— se utilizan con fi nes de investigación y establecen tres subtipos clínicos de la enfermedad: una variante frontal, donde predominan los cambios comportamentales; la demencia semántica, caracterizada por una afasia fl uente con pérdida del conocimiento semántico, y la afasia primaria progresiva.
Los síntomas iniciales de la demencia frontotemporal están relacionados con cambios de personalidad, así como con alteraciones comportamentales, del afecto, del lenguaje o de las funciones ejecutivas (23- 25). Sin embargo, son variables de acuerdo con el área cortical afectada: puede existir desinhibición, probablemente secundaria al daño de la corteza basal frontal y temporal anterior, y es más frecuente en pacientes con disfunción frontotemporal derecha (26-27). Las alteraciones del lenguaje son frecuentes, en especial en pacientes con una afectación asimétrica izquierda (28). La apatía se asocia con disfunción del cíngulo anterior (26).
La insuficiencia en las funciones ejecutivas es una característica de la variante frontal, con difi cultades en la planeación y organización del espacio. Son frecuentes la hiperoralidad, el aumento de peso y los cambios en la preferencia de los alimentos, así como las compulsiones en relación con la alimentación; igualmente, pueden ser secundarios a disfunción frontotemporal y cambios en las concentraciones de serotonina o dopamina (26).
En algunos pacientes se observa un aumento en el interés en la música y las artes visuales, asociado con creatividad musical o visual, en especial en casos de una afectación focal del lóbulo temporal anterior izquierdo (29). Los hallazgos en la evaluación neuropsicológica son útiles en el diagnóstico diferencial de la demencia frontotemporal (30).
La mayoría de pacientes, incluso en etapas tempranas de la enfermedad, presentan una afectación importante de las funciones ejecutivas, el lenguaje y la memoria de trabajo. Por lo general, están preservadas las habilidades visuoconstruccionales, a diferencia de la enfermedad de Alzheimer (1). La evaluación neuropsicológica permite, además, hacer un diagnóstico diferencial entre las variantes de la demencia frontotemporal (3).
Variante frontal de la demencia frontotemporal
La variante frontal representa el 90% de los casos de demencia frontotemporal y su manifestación son los cambios de comportamiento (9, 31- 33), de los cuales los más frecuentes son pérdida de emotividad, desinhibición, disminución del cuidado personal, pérdida de interés en las actividades que antes realizaba, aumento en el consumo de alimentos con una preferencia por alimentos dulces y comportamientos motores estereotipados (31,34).
Los criterios diagnósticos de la variante frontal incluyen trastornos comportamentales de inicio insidioso, con una progresión lenta, deterioro social, signos precoces de desinhibición, rigidez e infl exibilidad mental, ausencia de conciencia de enfermedad (anosognosia) y trastornos afectivos y del lenguaje (Tabla 2). Se han descrito dos sín-dromes clínicos de la variante frontal (31):
• Apático: mediado por atrofia del área dorsolateral del lóbulo frontal.
• Desinhibido: mediado por atrofi a del área orbitomedial del lóbulo frontal y el polo temporal.
En los dos síndromes, se conservan las actividades instrumentales, el lenguaje y las funciones visuoespaciales (31). La memoria puede llegar a afectarse, pero no existe una franca amnesia al inicio de la enfermedad. En algunas ocasiones se presenta una difi cultad en generar información, con implicaciones en la abstracción y la organización (9,35).
Demencia semántica
La demencia semántica se caracteriza por una pérdida del signifi cado de las palabras, aun cuando se conservan los aspec tos fonológicos y sintácticos del lenguaje (Tabla 3). Al comienzo, los pacientes tienen poca conciencia de su defecto y pierden, de manera progresiva, su capacidad para entender el signifi cado de las palabras, así como estímulos visuales, olores, sabores y sonidos no verbales (31). Sin embargo, pueden emplear los objetos que dicen no saber qué son.
La demencia semántica es secundaria a una degeneración de la región anterior de los lóbulos temporales, y tanto los estudios en imágenes como en autopsias han demostrado una afectación importante de los giros temporales inferior y medio (31,36).
Aunque el défi cit semántico es predominante en el cuadro clínico, las personas pueden presentar cambios comportamentales, que son diferentes a los que se evidenciados en la variante frontal. Los pacientes con demencia semántica ocupan gran parte de su tiempo en una actividad única como la pintura, lo que contrasta con la pérdida de interés en aquellos con demencia frontotemporal variante frontal.
También pueden presentar comportamientos hipocondriacos o histriónicos, a diferencia de la variante frontal, donde se observa una disminución de la reactividad a los diferentes estímulos (31). Tanto en la demencia semántica como en la variante frontal puede verse un aumento en el apetito, con preferencia por los alimentos dulces (34). Las funciones visuoespaciales están intactas al inicio de la enfermedad (9,37-38).
En algunos pacientes con demencia semántica, se desarrolla un nuevo interés en el arte: hacen pinturas realistas o surrealistas sin un signifi cado simbólico o un componente abstracto. Los dibujos son realizados de manera compulsiva y, a veces, son repetidos varias veces. Los colores utilizados son, por lo general, morado, amarillo o azul (39).
Los mecanismos por medios de los cuales se produce la creatividad en estos pacientes no se conocen con exactitud. La corteza parietotemporal posterior no se ve afectada en la demencia semántica, por lo que no se perturban las actividades visuoespaciales, a diferencia de lo que ocurre en pacientes con enfermedad de Alzheimer. Se ha sugerido que la necesidad compulsiva de pintar también ayuda a desarrollar la creatividad visual, y la repetición constante lleva a la perfección del arte. Por otro lado, en la demencia semántica hay un hipometabolismo constante en el lóbulo temporal anterior izquierdo, y esto puede llevar a una “liberación” del hemisferio derecho, orientado visualmente, y por lo tanto, un aumento en la creatividad artística visual (39). Es menos frecuente que la variante frontal y la historia familiar puede estar presente en 25% de los casos (9).
Afasia primaria progresiva
Los pacientes con afasia primaria progresiva usualmente consultan por difi cultades para la nominación; anormalidades en el discurso, con disminución en el tamaño de las frases; parafasias fonológicas; alteraciones en la velocidad del lenguaje, la articulación y la prosodia, y errores gramaticales (40-42).
El cuadro clínico puede variar de acuerdo con la afectación cortical: algunos pacientes tienen difi cultades para encontrar las palabras y expresar lo que piensan; otros, para entender el signifi cado de las palabras, y otros tantos, en la nominación de objetos (40,43). Se admite la existencia de dos variantes de este síndrome afásico: fl uente y no fl uente. En la variante fl uente, generalmente se ve afectado el procesamiento semántico, pero la fonología y la sintaxis se conservan. En la no fl uente, las alteraciones del lenguaje se asemejan a la afasia de Broca, con errores gramaticales y parafasias fonológicas. Sin embargo, son características en este síndrome la relativa conservación de la comprensión, de la inteligencia no verbal y de la memoria (40,43,44).
En pacientes con enfermedad de Alzheimer, la afectación del lenguaje produce una afasia que, por lo general, es fl uente; mientras en la afasia primaria progresiva puede ser no fl uente (40,45). El signo más frecuente en afasia primaria progresiva es la anomia (incapacidad para encontrar las palabras correctas en una conversación, o nombrar objetos), y muchos pacientes permanecen en una fase anómica durante la mayor parte de la enfermedad, con una progresión gradual del défi cit, que puede llegar incluso al mutismo. En algunos pacientes, sin embargo, pueden observarse distintas formas de agramatismo o difi cultades en la comprensión del lenguaje (40).
La comprensión escrita frecuentemente es buena. La lectura en voz alta se encuentra alterada, mientras la escritura está preservada, aunque puede existir disortografía, agramatismo o fallas de sintaxis. En las fases iniciales e intermedias del proceso, las personas utilizan recursos no verbales, esto es, señalan con el dedo o hacen mímica; pero en las fases avanzadas hay una tendencia creciente al mutismo y a la pérdida de la iniciativa para la comunicación no verbal (9, 46,47).
En fases avanzadas de la enfermedad, cuando todos los aspectos del lenguaje están gravemente afectados, puede encontrarse también défi cit de memoria y cambios comportamentales (40). El diagnóstico se hace cuando hay un daño en el lenguaje, mientras que otras áreas de la esfera cognoscitiva, como memoria, gnosias y praxias, están prácticamente intactas; cuando existe una afectación aislada del lenguaje durante dos años, y cuando los estudios de imágenes estructurales no revelan una lesión específi ca, diferente a la atrofi a, que pueda producir el défi cit del lenguaje (Tabla 4) (40,41,48).

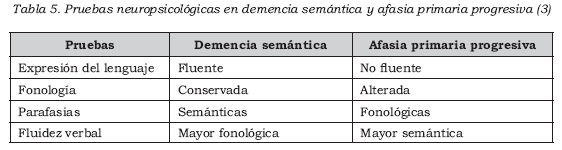
Las pruebas neuropsicológicas son útiles para hacer un diagnóstico temprano (Tabla 5) (3,40,49). Sin embargo, debido a que la mayoría de pruebas requieren instrucciones verbales, respuestas verbales o un razonamiento verbal, ocasionalmente hay conclusiones erróneas respecto a la afectación de otras áreas diferentes al lenguaje (40).
Otras variantes
Demencia frontal con parkinsonismo
La demencia frontal con parkinsonismo es un síndrome clínico, caracterizado por la presencia de cambios comportamentales, demencia y parkinsonismo, asociado con alteraciones en el cromosoma 17, con una herencia autosómica dominante. Se ha demostrado la presencia de mutaciones en el gen que codifi ca para la proteína tau, asociada con microtúbulos, localizado por lo general en el brazo largo de este cromosoma: 17q21-22 (1).
Demencia frontotemporal asociada con enfermedad de motoneurona Al evaluar a un paciente con diagnóstico de demencia frontotemporal, el médico siempre debe estar atento ante la posibilidad de una asociación con enfermedad de motoneurona (1,50), ya que se ha demostrado que, por lo menos, 15% de los pacientes con demencia frontotemporal presentan además esclerosis lateral amiotrófi ca, y 20% de los pacientes con esclerosis lateral amiotrófi ca, síntomas y signos de demencia frontotemporal (51).
Las alteraciones motoras pueden preceder, coincidir o seguir a los cambios comportamentales y cognoscitivos. Aunque los pacientes que presentan este síndrome clínico, por lo general, no presentan el curso clínico rápidamente progresivo de la esclerosis lateral amiotrófi ca, el deterioro clínico es más acelerado que en otros subtipos de demencia frontotemporal, y puede llevar a la muerte por disfagia progresiva o falla respiratoria (1).
Degeneración corticobasal
La degeneración corticobasal es un síndrome clínico caracterizado por rigidez asimétrica, apraxia, alteraciones visuoconstruccionales, fenómeno de mano extraña y mioclonías, secundario a afectación de los lóbulos frontales, parietales y ganglios basales (52-53). Esta degeneración tiene hallazgos patológicos comunes con la demencia frontotemporal —entre estos, pérdida neuronal, gliosis, presencia de inclusiones neuronales y ausencia de placas de amiloide y ovillos neurofi brilares— (54).
Imágenes cerebrales
Es necesario realizar un estudio imagenológico, con resonancia magnética cerebral o imágenes funcionales, en todos los pacientes, para excluir lesiones que generan síntomas que sugieren una demencia frontotemporal. Un tumor de línea media, de lento crecimiento, puede producir cambios comportamentales sutiles que simulan una demencia frontotemporal, sin otros signos neurológicos focales (1).
Resonancia magnética cerebral
La resonancia se observa atrofi a cortical (que es más prominente en los lóbulos temporales y frontales), dilatación ventricular y atrofi a de la cabeza del núcleo caudado. La atrofi a puede ser asimétrica, pues esta es una característica en los casos de afasia primaria progresiva y demencia semántica. El análisis volumétrico de diferentes regiones corticales puede ser útil en el diagnóstico diferencia con enfermedad de Alzheimer (55-57). Adicionalmente, hay hallazgos característicos en algunas formas de demencia frontotemporal (55, 58-60):
• Variante frontal: atrofi a dorsolateral frontal derecha, atrofi a del cíngulo y de la corteza insular anterior en la variante frontal.
• Afasia primaria progresiva: atrofi a perisilviana izquierda y dorsolateral frontal en la afasia primaria progresiva.
• Demencia semántica: atrofi a de la amígdala, corteza orbitofrontal, temporal anterior e insular anterior.
Imágenes funcionales
Las imágenes funcionales son útiles en el diagnóstico en las fases tempranas de la enfermedad, cuando la atrofi a aún no es evidente. Se observa hipoperfusión en las imágenes de tomografía computarizada por emisión de fotón único (SPECT, por su sigla en inglés) o hipometabolismo en la tomografía por emisión de positrones (PET, por su sigla en inglés), en los lóbulos frontales y temporales (56,61); así como disminución de la activación cortical frontal y parietal en la resonancia magnética funcional (62). En imágenes de perfusión por resonancia magnética se evidencia una disminución de la perfusión en los lóbulos frontales y temporales, de una forma similar al SPECT y al PET (63).
Patología
A diferencia de lo que ocurre en la enfermedad de Alzheimer, las cortezas parietal posterior, temporal posterior y occipital están relativamente conservadas. Posiblemente, las primeras áreas corticales afectadas son el cíngulo anterior, la región insular anterior y las regiones frontales ventrales (64).
Macroscópicamente se observa atrofi a del hipocampo y de los núcleos basales (en especial la cabeza del núcleo caudado), ensanchamiento de los ventrículos, palidez de la sustancia nigra y, en algunos casos, atrofi a de los cordones laterales de la médula (1). La utilización de técnicas de microscopia convencional permite identifi car una pérdida neuronal en la corteza atrófi ca, microvacuolización, gliosis astrocítica en la capa II de la corteza cerebral y, en algunos casos, neuronas abalonadas (1).
La neuropatología de la demencia frontotemporal puede ser clasifi - cada de acuerdo con la presencia de inclusiones neuronales y gliales (65): tau positiva, ubiquitina positiva tau negativa y sin inclusiones. La proteína tau, codifi cada por el cromosoma 17, está compuesta por seis isoformas. La mitad de las isoformas tienen tres secuencias repetitivas con sitios de unión a microtúbulos y se han denominado 3R, mientras que la otra mitad de isoformas presenta cuatro secuencias repetitivas y se han denominado 4R. Las isoformas 4R son características de la degeneración corticobasal y la parálisis supranuclear progresiva, mientras que las isoformas 3R son características de la demencia frontotemporal (1,66).
Las inclusiones de proteína tau, de cualquier tipo, son infrecuentes en estudios post mórtem, y se presentan principalmente en casos de afasia primaria progresiva y degeneración corticobasal (67). Aproximadamente, 50% de los casos de demencia frontotemporal presentan inclusiones, ubiquitinas positivas y tau negativas; una tercera parte, inclusiones tau positivas, y 15% no tiene inclusiones (1). La presencia de inclusiones citoplasmáticas de ubiquitina debe hacer sospechar una asociación con enfermedad de motoneurona (1,66).
Tratamiento
En la actualidad no existe un tratamiento farmacológico específi co para la demencia frontotemporal. Se han utilizado en algunos casos inhibidores de colinesterasa, pero no se ha demostrado su efi cacia (1), y pueden aumentar la irritabilidad (68). Las intervenciones farmacológicas para el manejo de las alteraciones comportamentales incluyen neurolépticos atípicos, antidepresivos, anticonvulsivos y agonistas de dopamina.
Estudios recientes con inhibidores de recaptación de serotonina y trazodona han demostrado su utilidad en el manejo de los cambios comportamentales, incluso en ausencia de síntomas depresivos (55). La desinhibición, la alteración en el control de impulsos y la agresión parecen estar en relación con una disminución en la transmisión serotoninérgica (69-70), al igual que las compulsiones y la preferencia por carbohidratos, lo que explicaría el efecto benéfico de los inhibidores de recaptación de serotonina en el manejo de estos síntomas (69,71).
La agresividad puede ser muy problemática para los pacientes y los cuidadores (72); en muchas situaciones, requiere una intervención farmacológica, en cuyo caso se prefi ere usar antipsicóticos atípicos (69,73), pero por corto tiempo, y debe considerarse el riesgo de ataque cerebro-vascular, en especial en pacientes con ataques previos de este tipo (74).
El manejo no farmacológico es muy importante en pacientes con diagnóstico de demencia frontotemporal. La educación a los pacientes y a los cuidadores acerca del carácter progresivo de la enfermedad es de vital importancia, al igual que lograr vínculos con programas de apoyo, como centros de hospital día o programas especiales de terapia ocupacional (72).
Conclusiones
La demencia frontotemporal, aunque es menos frecuente que la enfermedad de Alzheimer, es responsable un porcentaje importante de las demencias degenerativas, ya que corresponde a 5%-7% de las series de autopsias y a 20% de los pacientes que mueren con demencia antes de los 70 años de edad. Se instaura, por lo general, durante la sexta década de la vida, y la edad de muerte es usualmente en la séptima década, aunque hay reportes de casos familiares en los cuales las primeras manifestaciones de la enfermedad se presentan durante la segunda década. En una minoría de los casos (<10%) hay una historia familiar. La mayoría de las formas familiares de demencia frontotemporal tienen una herencia autosómica dominante y se asocian con mutaciones en el gen que codifi ca la proteína tau (asociada con microtúbulos), localizado en el cromosoma 17.
Los síntomas iniciales de la demencia frontotemporal están relacionados con cambios de personalidad, así como alteraciones comportamentales, del afecto, el lenguaje o las funciones ejecutivas. Sin embargo, son variables, de acuerdo con el área cortical afectada: la variante frontal representa el 90% de los casos de demencia frontotemporal y su manifestación son los cambios de comportamiento.
La demencia semántica se caracteriza por una pérdida del signifi cado de las palabras, aun cuando se conservan los aspec tos fonológicos y sintácticos del lenguaje. Los pacientes con afasia primaria progresiva usualmente consultan por difi cultades para la nominación; anormalidades en el discurso, con disminución en el tamaño de las frases; parafasias fonológicas; alteraciones en la velocidad del lenguaje, la articulación y la prosodia, y errores gramaticales.
Las intervenciones farmacológicas para el manejo de las alteraciones comportamentales incluyen neurolépticos atípicos, antidepresivos, anticonvulsivos y agonistas de dopamina. Estudios recientes con inhibidores de recaptación de serotonina y trazodona han demostrado utilidad de estos medicamentos en el manejo de los cambios comportamentales, incluso en ausencia de síntomas depresivos. El manejo no farmacológico es muy importante en pacientes con diagnóstico de demencia frontotemporal.
Referencias
1. Graff-Radford N, Woodruff B. Frontotemporal dementia. Semin Neurol. 2007 Feb;27(1):48-57. [ Links ]
2. Ikeda M, Ishikawa T, Tanabe H. Epidemiology of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2004;17:265-8. [ Links ]
3. Montañés P, Cano C, Pedraza O, Peñalosa M, Rubiano LD, Gamez A, et al. Demencia no Alzheimer: variante frontal de la demencia fronto-temporal. Revista de la Asociación Colombiana de Gerontología y Geriatriatría. 2003;17(4):539-83. [ Links ]
4. Greene J, Patterson K, Xuereb J, Hodges JR. Alzheimer disease and non-fluent progressive aphasia. Arch Neurol. 1996;53:1072-8. [ Links ]
5. Snowden J, Neary D, Mann DMA, Goulding PJ, Testa HJ. Progressive language disorder due to lobar atrophy. Ann Neurol. 1992; 31:174-83. [ Links ]
6. Hodges JR, Patterson K, Ward R, Garrard P, Bak T, Perry R, et al. The differentiation of semantic dementia and frontal lobe dementia (temporal and frontal variants of frontotemporal dementia) from early Alzheimer disease: a comparative neuropsychological study. Neuropsychology. 1999;13:31-40. [ Links ]
7. Montañés P, Matallana D. Fragmentación de la memoria en una paciente con demencia semántica. Neurociencias en Colombia. 2001;9:53-60. [ Links ]
8. Kertesz A, Munoz D. Pick’s disease, frontotemporal dementia, and Pick complex: emerging concepts. Arch Neurol. 1998;55:302-4. [ Links ]
9. Cano CA, Ramírez RA. Avances nosológicos de las demencias. Caracterización de los pacientes con demencia frontotemporal. Med UNAB. 2004;7:84-8. [ Links ]
10. Hodges JR, Miller B. The classification, genetics and neuropathology of frontotemporal dementia. Neurocase. 2001;7:31-5. [ Links ]
11. Johnson JK, Diehl J, Mendez MF, Neuhaus J, Shapira JS, Forman M, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925-30. [ Links ]
12. Roberson ED, Hesse JH, Rose KD, Slama H, Johnson JK, Yaffe K, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005;65:719-25. [ Links ]
13. Wilhelmsen K, Forman MS, Rosen HJ, Alving LI, Goldlman J, Feiger J, et al. 17q linked FTDALS without tau mutations with tau and a-synuclein inclusions. Arch Neurol. 2004;61:398-406. [ Links ]
14. Poorkaj P, Bird T, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815-25. [ Links ]
15. Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, et al. Tau gene mutation in familial progressive subcortical gliosis. Nat Med. 1999;5:454-7. [ Links ]
16. Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol. 2000;48:939-43. [ Links ]
17. Hosler BA, Siddique T, Sapp PC, Huang W, Huang MC, Hussain A, et al. Linkage of familial amyotrophic lateral sclerosis with front frontotemporal dementia to chromosome 9q21-q22. Jama. 2000;284:1664-9. [ Links ]
18. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806-8. [ Links ]
19. Dermaut B, Kumar-Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J, et al. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55:617-26. [ Links ]
20. Geschwind D, Karrim J, Koras N, Nelson SF, Miller BL. The Apo E4 allele is not a significant risk factor for frontotemporal dementia. Ann Neurol. 1998;44:134-8. [ Links ]
21. Mesulam M-M, Johnson N, Grujic Z, Weintraub S. Apolipoprotein E genotypes in primary progressive aphasia. Neurology. 1997;49:51-5. [ Links ]
22. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546-52. [ Links ]
23. Lindau M, Almkvist O, Kushi J, Boone K, Johansson SE, Wahlund LO, et al. First symptoms: frontotemporal dementia versus Alzheimer’s disease. Dementia & Geriatri Cog Disord. 2000;11:286-93. [ Links ]
24. Seeley WS, Bauer A, Miller BL, Gorno- Tempini ML, Kramer JH, Weiner M, Rosen HJ. The natural history of temporal variant frontotemporal dementia. Neurology. 2005;64:1384-90. [ Links ]
25. Miller BL, Darby AL, Swartz JR, Yener GG, Mena I. Dietary changes, compulsions and sexual behavior in frontotemporal degeneration. Dementia. 1995;6:195-9. [ Links ]
26. Rosen HJ, Allison S, Schauer GF, Gorno-Tempini ML, Weiner MW, Miller BL. Neuroanatomical correlates of behavioral disorders in dementia. Brain. 2005;128:2612-25. [ Links ]
27. Liu W, Miller BL, Kramer J, Rankin K, Wyss-Coray C, Gearhart R, et al. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology. 2004; 62:742-8. [ Links ]
28. Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335-46. [ Links ]
29. Miller BL, Cummings JL, Boone K, Prince F, Ponton M, Cotman C. Emergence of artistic talent in frontotemporal dementia. Neurology. 1998;51:978-81. [ Links ]
30. Diehl J, Monsch AU, Aebi C, Wagenpfeil S, Krapp S, Grimmer T, et al. Frontotemporal dementia, semantic dementia, and Alzheimer’s disease: the contribution of standard neuropsychological tests to differential diagnosis. J Geriatr Psychiatry Neurol. 2005;18:39-44. [ Links ]
31. Snowden JS, Bathgate D, Varma A, Blackshaw A, Gibbons ZC, Neary D. Distinct behavioural profiles in frontotemporal dementia and semantic dementia. J Neurol Neurosurg Psychiatry. 2001;70;323-32. [ Links ]
32. Miller BL, Cummings JL, Villanueva- Meyer J, Boone K, Mehringer CM, Lesser IM, et al. Frontal lobe degeneration: clinical, neuropsychological and SPECT characteristics. Neurology. 1991;41:1374-82. [ Links ]
33. Gustafson L. Clinical picture of frontal lobe degeneration of non-Alzheimer type. Dementia. 1993;4:143-8. [ Links ]
34. Ikeda M, Brown J, Holland AJ, Fukuhara R, Hodges JR. Changes in appetite, food preference, and eating habits in frontotemporal dementia and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;73;371-6. [ Links ]
35. Gustafson L. Frontal lobe degeneration non-Alzheimer type II: clinical picture and differential diagnosis. Archiv Gerontol Geriatr. 1987;6:209-23. [ Links ]
36. Mummery CJ, Patterson K, Price CJ. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol. 2000;47:36-45. [ Links ]
37. Hodges JR, Patterson K. Semantic dementia-progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115:1783-806. [ Links ]
38. Snowden J, Griffiths H. Semantic-episodic memory interactions in semantic dementia: implications for retrograde memory function. Cognitiv Neuropsychol. 1996;13:1101-37. [ Links ]
39. Miller BL, Craig EH. Portraits of artists: emergence of visual creativity in dementia. Arch Neurol. 2004;61:842-4. [ Links ]
40. Mesulam M-M. Primary progressive aphasia: a language-based dementia. N Engl J Med. 2003;349:1535-42. [ Links ]
41. Méndez MF, Clark DG, Shapira JS, Cummings JL. Speech and language in progressive nonfluent aphasia compared with early Alzheimer’s disease. Neurology. 2003;61:1108-13. [ Links ]
42. Krefft TA, Graff-Radford NR, Dickson DW, Baker M, Castellani R. Familial primary progressive aphasia. Alzheimer Dis Assoc Disord. 2003;17:106-12. [ Links ]
43. Snowden JS, Neary D, Mann DMA, Goulding PJ, Testa HJ. Progressive language disorder due to lobar atrophy. Ann Neurol. 1992;31:174-83. [ Links ]
44. Montañés P, Matallana D, García R, Cano C. Deterioro selectivo del lenguaje debido a degeneración temporal focal: análisis comparativo entre un caso de afasia primaria progresiva y un caso de demencia semántica. Univ Med. 2001;42:119-30. [ Links ]
45. Price BH, Gurvit H, Weintraub S, Geula C, Leimkuhler E, Mesulam M. Neuropsychological patterns and language deficits in 20 consecutive cases of autopsy- confirmed Alzheimer’s disease. Arch Neurol. 1993;50:931-7. [ Links ]
46. Poeck K, Luzzatti C. Slowly progressive aphasia in three patients: the problem of accompanying neuropsychological deficit. Brain. 1988;111:151-68. [ Links ]
47. Graff-Radford N, Damasio A, Hyman MN, Hart D, Tranel H, Damasio G, et al. Progressive aphasia in patients with Pick’s disease. Neurology. 1990;40:620-6. [ Links ] 48. Mesulam M-M. Primary progressive aphasia. Ann Neurol. 2001;49:425-32. [ Links ]
49. Weintraub S, Rubin NP, Mesulam MM. Primary progressive aphasia: longitudinal course, neuropsychological profile, and language features. Arch Neurol. 1990;47:1329-35. [ Links ]
50. Strong MJ, Lomen-Hoerth C, Caselli RJ. Cognitive impairment, frontotemporal dementia, and the motor neuron diseases. Ann Neurol. 2003;54(Suppl 5):S20-S23. [ Links ]
51. Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller BL. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094-97. [ Links ]
52. Bergeron C, Pollanen MS, Weyer L, Black SE, Lang AE. Unusual clinical presentations of cortical-basal ganglionic degeneration. Ann Neurol. 1996;40:893-9. [ Links ]
53. Kertesz A, Martínez-Lage P, Davidson W, Munoz DG. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000;55:1368-75. [ Links ]
54. Jackson M, Lowe J. The new neuropathology of degenerative frontotemporal dementias. Acta Neuropathol. 1996;91:127-34. [ Links ]
55. Pasquier F, Fukui T, Sarazin M. Laboratory investigations and treatment in frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S32-S35. [ Links ]
56. Laakso M, Frisoni GB, Könönen M, Mikkonen M, Beltramello A, Geroldi C, et al. Hippocampus and entorhinal cortex in frontotemporal dementia and Alzheimer’s disease: a morphometric MRI study. Biol Psychiatry. 2000;47:1056-63. [ Links ]
57. Whitwell JL, Josephs KA, Rossor MN, Stevens JM, Revesz T, Holton JL, et al. Magnetic resonance imaging signatures of tissue pathology in frontotemporal dementia. Arch Neurol. 2005;62:1402-08. [ Links ]
58. Rosen HJ, Hartikainen KM, Jagust W, Kramer JH, Reed BR, Cummings JL, et al. Utility of clinical criteria in differentiating frontotemporal lobar degeneration (FTLD), from AD. Neurology. 2002;58:1608-15. [ Links ]
59. Galton CJ, Patterson K, Graham K, Lambon-Ralph MA, Williams G, Antoun N, et al. Differing patterns of temporal atrophy in Alzheimer’s disease and semantic dementia. Neurology. 2001;24;57:216-25. [ Links ]
60. Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, et al. Common and differing patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198-208. [ Links ]
61. McMurtray AM, Chen AK, Shapira JS, Chow TW, Mishkin F, Miller BL, Mendez MF. Variations in regional SPECT hypoperfusion and clinical features in frontotemporal dementia. Neurology. 2006;66:517-22. [ Links ]
62. Rombouts SA, Van Swieten JC, Pijnenburg YAL, Goekoop R, Barkhof F, Scheltens P. Loss of frontal fMRI activation in early frontotemporal dementia compared to early AD. Neurology. 2003;60:1904-8. [ Links ]
63. Du AT, Jahng GH, Hayasaka S, Kramer JH, Rosen HJ, Gorno-Tempini ML, et al. Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology. 2006;67:1215-20. [ Links ]
64. Brun A. Frontal lobe dementia of the non-alzheimer type revisited. Dementia. 1993;4:126-31. [ Links ]
65. McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of work group on frontotemporal dementia and Pick’s disease. Arch Neurol. 2001;58:1803-9. [ Links ]
66. Muñoz DG, Dickson DW, Bergeron C. The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S24-S28. [ Links ]
67. Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996-2005. [ Links ]
68. Perry R. Miller BL. Pharmacological treatment of frontotemporal dementia. Int J Geriat Psychiatry. 2000;2:127- 31. [ Links ]
69. Perry R, Miller BL. Behavior and treatment in frontotemporal dementia. Neurology. 2001;56(Suppl 4):S46-S51. [ Links ]
70. Roy A, Linnoila M. Suicidal behavior, impulsiveness and serotonin. Acta Psychiatr Scand. 1988;78:529-35. [ Links ]
71. Sparks DL, Markesbery WR. Altered serotonergic and cholinergic synaptic markers in Pick’s disease. Arch Neurol. 1991;48:796-9. [ Links ]
72. Mourik JC, Rosso SM, Niermeijer MF, Duivenvoorden HJ, Van Swieten JC, Tibben A. Frontotemporal dementia: behavioral symptoms and caregiver distress. Dement Geriatr Cogn Disord. 2004;18:299-306. [ Links ]
73. Fava M. Psychopharmacologic treatment of pathologic aggression. Psychiatr Clin North Am. 1997;20:427- 51. [ Links ]
74. Bullok R. Treatment of behavioural and psychiatric symptoms in dementia: implications of recent safety warnings. Current Medical Research and Opinión. 2005;21:1-10. [ Links ]
Recibido para evaluación: 2 de marzo de 2007 Aprobado para publicación: 1 de junio de 2007

