










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Psiquiatría
Print version ISSN 0034-7450
rev.colomb.psiquiatr. vol.37 no.1 Bogotá Jan./Mar. 2008
Genética de la demencia frontotemporal
Genetics of Frontotemporal Dementia
Jorge Luis Granadillo de Luque1 Ignacio Zarante2
1 Médico interno, Universidad Industrial de Santander, Bucaramanga, Colombia.
2 Médico genetista. Profesor asociado del Instituto de Genética Humana, Facultad de Medicina, Pontificia Universidad Javeriana, Bogotá, Colombia. izarante@javeriana.edu.co
Resumen
Introducción: La demencia frontotemporal (DFT) es una enfermedad neurodegenerativa, caracterizada por la atrofia circunscrita de los lóbulos frontales y temporales. Se acompaña de cambios en el comportamiento o alteraciones del lenguaje. Su inicio es insidioso, y su curso, progresivo. Objetivo: Revisar de manera general la DFT, haciendo hincapié en sus bases genéticas. Métodos: Revisión de la literatura médica actual sobre el tema. Resultados: Últimamente se han estudiado familias con herencia autosómica dominante de esta enfermedad, entre las cuales se han hallado diferentes genes causales. De estos los más importantes son el gen de la proteína asociada con los microtúbulos tau y el gen de la progranulina. Conclusión: Es necesario considerar un abordaje genético en el momento de enfrentarse a un paciente con DFT, para así indagar profundamente en los antecedentes familiares, con el fin de brindar una adecuada asesoría genética que permita aclarar las principales dudas de los pacientes y sus familias sobre este aspecto.
Palabras clave: demencia, genética, proteínas tau, progranulina.
Abstract
Introduction: Frontotemporal dementia (FTD) is a neurodegenerative disease characterized by the circumscribed atrophy of the frontal and temporal lobes along with progressive and insidious changes in behaviour and/or language impairment. Objective: To review FTD, emphasizing its genetic bases. Methods: Review of the current medical literature about the subject. Results: Families with a dominant autosomic inheritance pattern of this disease have been recently studied and different causal genes have been found, the most important being the microtubule associated protein tau gene and the progranulin gene, among others. Conclusions: It is important to consider a genetic approach in the assessment of a patient with FTD, investigating deeply into his family background. This way a proper genetic counseling can be performed thus solving the main doubts that the patient and relatives may have.
Key words: Dementia, genetics, tau proteins, progranulin.
Introducción
Hace más de un siglo, Arnold Pick describió unos síndromes focalizados, asociados con la atrofia circunscrita de los lóbulos frontales y temporales. Luego Alzheimer describió los cuerpos de inclusión intracelulares, a los cuales posteriormente se les dio el nombre de cuerpos de Pick, y se encontró que éstos se relacionaban con la atrofia circunscrita. De esta manera, a todo cuadro clínico caracterizado por desinhibición y atrofia frontal se le consideraba enfermedad de Pick, expresión que progresivamente fue cayendo en desuso, debido a que la mayoría de atrofias lobares no presenta cuerpos de Pick, por lo cual actualmente se prefiere la expresión demencia frontotemporal (DFT) (1-3).
Recientemente, grandes avances en el área nos han permitido tener un mayor conocimiento acerca de la etiología y las bases genéticas de esta enfermedad. El médico psiquiatra debe tener un conocimiento sólido en esta área, pues es él quien maneja pacientes con síndromes cognoscitivos a los cuales, además de diagnosticar y tratar adecuadamente, debe brindarles la información necesaria, tanto al paciente como a sus familiares, acerca de las causas de la enfermedad, su heredabilidad y las perspectivas de manejo.
Definición
La DFT es una enfermedad neurodegenerativa poco frecuente, la cual, más que una enfermedad por sí sola, constituye un complejo de enfermedades, cuya característica común es la atrofia circunscrita de los lóbulos frontales o temporales, ya sea simétrica o asimétrica, lo cual determina la aparición de tres cuadros diferentes: uno con predominio de síntomas comportamentales —variante comportamental de la demencia frontotemporal (DFTc), denominado en inglés FTD-bv— y otros dos con predominio de deficiencias en el área del lenguaje —afasia progresiva primaria (APP) y la demencia semántica (DS)— (4), los cuales describimos a continuación:
Variante comportamental de la demencia frontotemporal
La DFTc presenta un inicio insidioso, caracterizado por un profundo cambio en la personalidad. Se presenta en la persona con un comportamiento desinhibido, impulsivo e inapropiado, o puede presentar abulia y pérdida de la iniciativa, con aplanamiento del ánimo. A diferencia de la enfermedad de Alzheimer y otros síndromes demenciales, en la DFTc existe una relativa preservación de la memoria, aunque puede haber quejas sobre alteraciones, algunas detectables en test formales de memoria, secundarias no a una amnesia primaria (hay conservación de la orientación y del recuerdo de eventos diarios), sino al déficit propio de la disfunción frontal.
Los déficits cognoscitivos se dan en las áreas de la atención, planeación, abstracción y resolu ción de problemas. Con el tiempo, el paciente presenta un detrimento importante en su capacidad de juicio, lo cual se refleja significativamente en el área financiera, ya que es frecuente que el individuo realice compras excesivas e impulsivas; además, puede darse un cambio en el patrón alimentario (como hiperfagia) y descuido en la higiene personal.
Igualmente, las personas con DFTc presentan conductas estereotipadas y repetitivas. Además, algunos pacientes pueden presentar comportamiento sexual inapropiado (4-6). Debido al inicio típico de este cuadro en pacientes de edad media y a los marcados cambios de personalidad y del comportamiento, muchas veces las personas afectadas son confundidas en sus fases tempranas con pacientes de tipo psiquiátrico (2).
A medida que progresa la enfermedad los trastornos cognoscitivos se hacen cada vez más prominentes, con alteración marcada del lenguaje hasta llegar al mutismo. En este punto el cuadro se hace difícil de diferenciar de la enfermedad de Alzheimer o cualquier otro tipo de demencias, por lo cual es importante indagar profundamente, durante la anamnesis, acerca de los signos y síntomas presentados durante el comienzo de este trastorno y su curso (5).
Afasia progresiva primaria
La APP fue inicialmente reportada por Mesulam, en 1982 (7). De inicio insidioso y curso progresivo, se caracteriza por alteraciones en el lenguaje, como dificultad para encontrar las palabras adecuadas durante una conversación, dificultad para darle nombre a los objetos (anomia), alteración en la comprensión de las palabras (habladas y escritas) y agramatismo.
Se presentan patrones del habla alterados, errores gramaticales prominentes, con eliminación de preposiciones, pronombres y conjunciones, lo cual resulta en un habla “telegráfica”. Por este motivo, a este trastorno también se le denomina afasia primaria no fluida (en contraposición a la afasia fluida de la demencia semántica, de la cual hablaremos más adelante), ya que el paciente presenta una disminución en el número de palabras habladas, y algunos pueden progresar hasta el mutismo. La presencia de una u otra deficiencia es variable: la anomia es, al parecer, la más frecuente y la comprensión de las palabras puede no estar tan afectada.
Las deficiencias en el lenguaje deben presentarse solas por, al menos, dos años. Es decir, no se debe acompañar de deficiencias mentales de otro tipo, ya sean de la memoria, del comportamiento, visuoespaciales, entre otras; además, se deben descartar causas secundarias de afasia (vasculares o tumores). Debido a que estos pacientes presentan una clara conciencia de su enfermedad, es relativamente frecuente que cursen con episodios de frustración y depresión; sin embargo, no se han reportado casos de suicidio. A medida que la enfermedad progresa, las deficiencias van más allá del área del lenguaje, y es mayor la afectación cognoscitiva, incluso pueden presentar comportamiento desinhibido y déficits motores (8-9).
Demencia semántica
A diferencia de la APP, en la DS se presenta una afasia fluida. Estos pacientes presentan problemas para dar nombre a los objetos y deterioro en la comprensión del lenguaje, pero conservan un discurso sin alteraciones cuantitativas o gramaticales, fluido, pero con un léxico pobre. En consecuencia, a medida que la enfermedad progresa, el discurso empeora en su contenido. Igualmente, los pacientes tienen dificultades en entender el signifi- cado de los rostros (prosopagnosia), objetos u otro tipo de estímulos sensitivos. Con frecuencia estos individuos no están conscientes de sus síntomas (3-6).
Se ha observado que estos tres subtipos de DFT pueden solaparse; es decir, un subtipo puede con el tiempo cumplir criterios para otro subtipo. Además, se ha visto que hay trastornos neurológicos que frecuentemente se pueden presentar en el contexto de una DFT, como el parkinsonismo, la degeneración corticobasal (DCB), la parálisis supranuclear progresiva (PSP) y la enfermedad de neurona motora (ENM) (2).
En un estudio, dos tercios de los pacientes con DFT presentaron durante su evolución un diagnóstico adicional de algún subtipo de DFT; además, encontraron que los pacientes con DS evolucionaban con mayor frecuencia hacia una DFTc que los demás subtipos. De la misma manera, la APP evolucionaba con mayor frecuencia hacia la DCB/PSP, que los demás subtipos y viceversa, lo cual sugiere una asociación entre estos síndromes (10).
Epidemiología
La DFT es la causa primaria más frecuente, después de la enfermedad de Alzheimer, de demencia presenil, y es responsable de aproximadamente el 20% de estas (1). Recientemente dos estudios han tratado este tema. El primer estudio, realizado en los Países Bajos, mostró una prevalencia de 3,6 por cada 100.000 personas en edades entre 50 y 59 años; 9,4 por cada 100.000 personas en edades entre los 60 y los 69, y 3,8 por cada 100.000 personas en edades entre los 70 y los 79 (11).
El otro estudio, realizado en Cambridgeshire, Reino Unido, reveló que la DFT constituye el 15,7% de todas las demencias de inicio temprano (<65 años), con una prevalencia de 15 (8,4-27,0) por cada 100.000 individuos con edades entre los 45 y los 65 años, valor similar a la prevalencia de enfermedad de Alzheimer de inicio temprano. Este mismo estudio mostró también una mayor prevalencia entre los hombres, con una razón de 14:3; sin embargo, esta aseveración escontraria al resultado de otros estudios, los cuales no han mostrado diferencias entre sexos, por lo cual se requieren más observaciones sobre este aspecto (12). No se encuentran estudios en la literatura médica actual que describan las características epidemiológicas de la DFT en Colombia o Hispanoamérica.
Diagnóstico
El diagnóstico de la DFT constituye uno de los aspectos más intrincados de esta patología, ya que es común que a cada uno de los subtipos ya mencionados se les den diferentes nombres, y aún que al mismo complejo de la DFT se le llame todavía enfermedad de Pick, entre otros términos. Existen diferentes criterios diagnósticos para la DFT, entre ellos encontramos los creados por Neary y cols., en 1998 (4), y los creados por Mckhann y cols., en el 2001 (Tabla 1) (5).

Neuropatología
Las características anatomopatológicas de la DFT son muy variadas y existen diferentes subtipos de hallazgos patológicos, los cuales no tienen una relación directa con los subtipos clínicos ya mencionados (10,13,14). Generalmente, los pacientes con DFT presentan marcada atrofia de los lóbulos frontales y temporales, la cual, como ya mencionamos, suele ser circunscrita (aunque en ocasiones puede afectar los lóbulos parietales) y simétrica o asimétrica.
Microscópicamente se observa pérdida neuronal, gliosis, espongiosis superficial, neuronas balonadas y células acromáticas (13,15). Los diferentes subtipos histopatológicos son:
• Cuerpos de Pick: se caracteriza por la presencia de los clásicos cuerpos de inclusión argirófilos, compuestos principalmente por la proteína asociada con los microtúbulos tau de tres repeticiones (3R-t); aunque en menor cuantía se puede encontrar proteína tau de cuatro repeticiones (4R-t) o una combinación de ambas isoformas. Estos cuerpos de inclusión se hallan principalmente en el giro dentado, las células piramidales del sector CA1 y el subiculum del hipocampo, al igual que en las capas II y VI de la neocorteza y en algunos núcleos subcorticales, como en el sistema estriado-palidonigral, el locus ceruleus y en el núcleo tuberal lateral del hipotálamo (13).
• Demencia frontotemporal tipo enfermedad de neurona motora (DFT-ENM): presenta inclusiones negativas para tau y sinucleína, pero positivas para ubiquitina (13). A pesar de su nombre, no se ha hallado correlación entre este tipo de patología y la presencia de síntomas de enfermedad de neurona motora (16-18). Estas inclusiones son igualmente positivas para p62 (17).
Clásicamente, estas inclusiones se encuentran en la capa II de la corteza cerebral frontal y en el giro dentado del hipocampo (19,20). Este patrón histopatológico es igualmente heterogéneo, y se divide en tipo I, que presenta inclusiones intranucleares, citoplasmáticas y placas neuríticas de ubiquitina; en el tipo II, donde se encuentran placas neuríticas de ubiquitina, pero pocas inclusiones citoplasmáticas, y en el tipo III, donde se hallan inclusiones citoplasmáticas, pero con pocas o ninguna placa neurítica. En las dos últimas no se encuentran inclusiones intranucleares (19, 21).
• Demencia frontotemporal sin histopatología distintiva (DTHD): aquella en la que no se hallan inclusiones mediante las técnicas actuales (13).
• DCB/PSP: debido al frecuente solapamiento clínico e histopatológico entre la DFT y la DCB/PSP, estos son incluidos en esta clasificación. También se ha propuesto la inclusión de la enfermedad por gránulos argirófilos en este grupo (5,13,22,23).
De acuerdo con la mayoría de los estudios, el subtipo más frecuente es la DFT-ENM (13,14,16). Según la isoforma de la proteína tau y el tipo de inclusiones que estén presentes, se pueden clasificar tal como se muestra en la Tabla 2 (13).

Exámenes paraclínicos
Pruebas neuropsicológicas
Con las pruebas neuropsicológicas se busca evaluar el cambio en el comportamiento en el paciente o las alteraciones del lenguaje, y comprobar una relativa preservación de las demás funciones cognoscitivas. Esto con el fin de diferenciar la DFT de los demás tipos de demencias.
Mediante el Inventario conductual frontal es posible determinar de manera cuantitativa las alteraciones comportamentales.
Este test aporta al diagnóstico, mientras que otras pruebas de tamizaje, como el Mini-Mental State Examination, son incapaces de diferenciar la DFT de la enfermedad de Alzheimer. La Western Aphasia Battery (WAB), entre otros, permite evaluar el lenguaje y el diagnóstico y clasificación de la afasia en los casos de APP y DS (10,25).
Neuroimagen
En la resonancia magnética, el hallazgo típico es la atrofia lobar, que es frontal y bilateral en la CFTC, y temporal izquierda en la APP. Se ha encontrado relación entre la atrofia lobar derecha y los síntomas comportamentales y entre la atro- fia perisilviana izquierda y la APP. Igualmente, puede verse un alargamiento de los ventrículos laterales y la atrofia del núcleo caudado.
Las regiones corticales afectadas pueden mostrar intensificación de la materia blanca subyacente en T2. Estos cambios pueden no verse al inicio de la enfermedad, por lo cual es recomendable usar neuroimagen funcional en estos casos, los cuales mostrarán hipoperfusión (en la tomografía por emisión de positrón único) o hipometabolismo (en la tomografía por emisión de positrones) en lóbulos temporales o frontales. La especificidad y sensibilidad de estos métodos no está determinada (2,26).
Electroencefalograma
Usualmente se encuentra dentro de parámetros normales, contrario a lo que sucede en la enfermedad de Alzheimer (27).
Líquido cefalorraquídeo (LCR)
Los estudios sobre las características del LCR son escasos. Se ha encontrado que las concentraciones del factor liberador de la corticotropina en el LCR están disminuidos en los pacientes con DFT, en comparación con aquellos con enfermedad de Alzheimer. A pesar de esto, no se han hallado unos marcadores capaces de diferenciar la DFT de otras patologías neurológicas. Se ha propuesto el estudio de las isoformas de la proteína tau (26).
Factores de riesgo ambientales
Realmente, pocos estudios han investigado los factores de riesgo relacionados con la DFT. Un estudio retrospectivo encontró una relación entre el trauma craneoencefálico y la DFT, además de hallar una asociación con la enfermedad tiroidea; sin embargo, esta última no fue estadísticamente significativa (28).
Genética de la DFT
Aunque la mayoría de los casos de DFT son esporádicos, se ha descrito entre un 20% y un 40% como familiares. En su mayoría presentan un patrón de herencia autosómico dominante (29,30). Dentro de las familias afectadas se han hallado diferentes genes implicados, como el gen de la proteína asociada con los microtúbulos tau (MAPT, por su sigla en inglés) (31) y el gen de la progranulina (PGRN) (32).
También existen familias con DFT ligada al cromosoma 9 (DFT-9) y al cromosoma 3 (CHMP2B), e incluso se han reportado mutaciones en el gen de la presenilina 1, gen que usualmente se ha relacionado con la enfermedad de Alzheimer (2). A continuación se describen los genes más destacados y su papel en la patogenia de la DFT (Tabla 3).
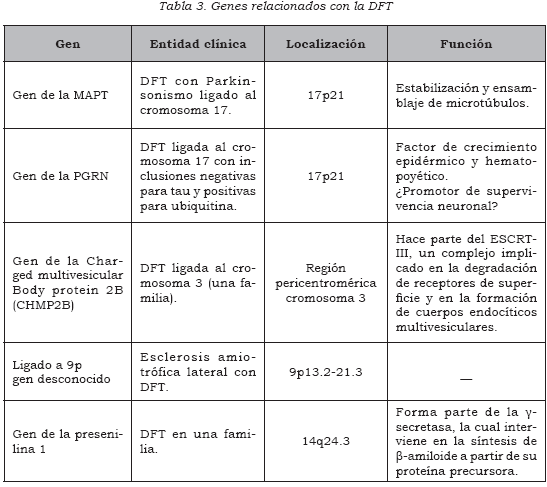
Gen de la MAPT
Existe un subgrupo de pacientes con DFT hereditaria de tipo autosómico dominante ligado a 17q21, que frecuentemente presenta síntomas parkinsonianos; por ello se le denomina a esta entidad demencia frontotemporal y parkinsonismo ligado al cromosoma 17 (DFTP-17) (29,33). En 1998 se encontró que el gen implicado en la DFTP-17 era el MAPT, que codifica para la proteína tau (31).
Tau pertenece a la familia de proteínas asociadas con los microtúbulos (MAP), y promueve el ensamblaje de los microtúbulos y su estabilización (34). Se encuentra normalmente en los axones, en los nucleolos, en los oligodendrocitos y en el músculo estriado (35). Esta proteína está codificada por un gen que posee 11 exones y genera 6 isoformas de la proteína tau mediante corte y empalme (splicing, en inglés) alternativo.
Cada proteína posee 3 o 4 repeticiones de 18 aminoácidos conocidos como dominios de unión al microtúbulo. Según el número de estos dominios que la isoforma posea, puede clasificarse como 4R-tau o de 3R-tau. La síntesis de una u otra isoforma está determinada por la inclusión o exclusión del exón 10 durante el corte y el empalme, ya que este exón codifica para el segundo dominio y la secuencia intercalada entre el primer y segundo dominio. Adicionalmente existe otro exón que está sometido a corte y empalme alternativo, que codifica para 237 aminoácidos localizados en el centro de la molécula; pero este se expresa en el sistema nervioso periférico y no es detectable en las neuronas de la corteza cerebral (31,34,35).
La 4R-tau tiene mayor afinidad por el microtúbulo que la 3R-tau. Esto puede no estar dado solamente por la diferencia entre el número de dominios, pues un experimento demostró que los dominios de unión al microtúbulo no actúan de manera independiente unos de otros, sino que la proteína tau posee un “dominio central de unión al microtúbulo”, compuesto por los dos primeros dominios y la secuencia que los separa (tanto en la 4R-tau como en la 3R-tau), que determina la afinidad de la tau al con el microtúbulo. Esto implica que existirían no solamente diferencias cuantitativas entre las dos isoformas, sino también cualitativas (34).
Hasta el momento se han reportado múltiples mutaciones en el gen de MAPT (36-52), las cuales pueden clasificar en dos tipos (53):
• Mutaciones codificantes: en este grupo se encuentran mutaciones de cambio de sentido y eliminaciones. Se caracterizan por afectar a las regiones codificantes del gen y alterar la secuencia de aminoácidos; por lo tanto, afectan la afinidad de tau con el microtúbulo y promueven su polimerización y precipitación. La más frecuente de estas es la P301L. Esta mutación ocurre en el exón 10, y se refiere solamente a la 4R-tau (ya que la 3R-tau no posee este exón). En los pacientes con dicha mutación usualmente se presentan inclusiones intraneuronales compuestas solamente por 4Rtau. Otras mutaciones afectan igualmente a las dos isoformas, como la G272V, cuyo lugar es en el exón 9 (31,53).
• Mutaciones que alteran el corte y empalme del exón 10: estas pueden ocurrir tanto dentro del exón 10 como en su intrón adyacente. Las primeras actúan al trastornar pequeñas secuencias que promueven o inhiben el corte y el empalme que se encuentran dentro del exón (ejemplo de estas son la N279K, delK280, N296N/H). Las segundas ocurren en el sitio de corte y empalme 5’ del exón 10, en la zona del intrón adyacente.
Se hipotetiza que en esta región normalmente se puede producir una estructura tipo stem-loop, que impide su reconocimiento y así el ARNm no es escindido en esa área, por lo que el exón 10 queda por fuera del ARNm maduro y se produce, en últimas, 3R-tau.
Este punto es importante para el mantenimiento de la razón 4Rtau/ 3R-tau dentro de niveles fisiológicos. Las mutaciones que ocurren en esta región producen una desestabilización de la estructura tipo stem-loop, con lo cual se promueve la inclusión del exón 10 y la mayor producción de 4R-tau, que eleva las cantidades de esta isoforma en el cerebro entre dos y seis veces. Ejemplos: +3, +11, +12, +13 (31,35,53,54). Este tipo de mutaciones tiene como resultado la alteración de la razón 4R-tau/3R tau, lo cual propicia la polimerización y precipitación de tau, con la consecuente formación de las ya descritas inclusiones intraneuronales (54).
De esta manera, el resultado inmediato de las mutaciones en el gen MAPT puede ser la alteración de la afinidad de tau por los microtúbulos. Esto los inestabiliza o altera la razón 4R-tau/3R-tau. Hoy en día no existe claridad acerca de los mecanismos mediante los cuales se producen las inclusiones de tau. Experimentos han mostrado que la fosforilación interviene en la regulación de la actividad de tau.
Las tau fosforiladas tienen menos afinidad por los microtúbulos y tienen mayor tendencia a unirse y precipitarse; igualmente, la fosforilación las hace menos susceptibles a la proteólisis y otro estudio encontró que la tau mutada (P301L) tiene menor afinidad con las fosforilasas. Todos estos hechos favorecen su precipitación. Otros procesos se encuentran posiblemente implicados, como la glicosilación, la ubiquitinación y la función de las chaperoninas. De todas maneras cabe resaltar que el aumento de las concentraciones de tau no mutada pueden, por sí solas, producir inclusiones y neurodegeneración (55).
Existe también controversia sobre el papel que desempeñan las inclusiones de tau en la patogenia de la DFT. Se afirma que la precipitación de tau puede ser un mecanismo de defensa que tiene como objetivo el secuestro de sustancias tóxicas (tau fosforilado, tal vez). Pero se discute esta propuesta y se señala que todas las neuronas que se degeneran presentan inclusiones y que el número de estas progresa a medida que avanza la enfermedad (55).
Como se mencionó, la DFTP-17 tiene un patrón de herencia autosómico dominante, por lo cual el individuo afectado tiene la posibilidad de transmitir el gen mutado en un 50%; sin embargo, la penetrancia es incompleta. Esto es necesario saberlo en el momento de realizar la asesoría genética (53).
Gen de la PGRN
Luego de haberse descubierto el papel de la MAPT en la DFTP-17, se encontró que ciertas familias presentaban una DFT de patrón autosómico dominante ligado a 17p21, que asociaban la enfermedad a la misma región cromosómica donde se encontraba el gen MAPT pero que, a pesar de esto, no presentaban mutaciones en ese gen. Además, presentaban un patrón histopatológico peculiar, en el cual se encontraban inclusiones negativas para tau y sinucleína, pero positivas para ubiquitina.
Estas inclusiones se localizaban no solamente en el citoplasma y como placas neuríticas, sino también intranuclearmente (DFT-ENM tipo I), lo cual es raro en la DFT esporádica (32, 56-58). Debido a que estas inclusiones son más características de aquellas enfermedades neurodegenerativas, cuya etiología es la expansión de poliglutaminas (como la enfermedad de Huntington o algunas ataxias hederitarias), se creyó posible que aquel DFT familiar estuviera dentro de este grupo; sin embargo, esto fue descartado posteriormente (56,59).
Por otro lado, se pensó que debía de haber mutaciones complejas del gen MAPT, sin detectarse; pero una vez descartado eso, se comenzaron a estudiar el resto de genes que se encontraban en aquella región (Cerca de 165). En el 2006 se halló que las mutaciones en la PGRN eran las responsables de la DFT en estas familias (32,60).
La PGRN es una proteína de 593 aminoácidos que posee siete repeticiones y media de un motivo de granulina de 12 cisteínas. Es precursora de unas pequeñas proteínas de 6 kDa llamadas granulinas, que ejercen complejos efectos modulatorios en el crecimiento celular. Hasta el momento se conocen 5 tipos en el ser humano. La PGRN se encuentra en todo el cuerpo, pero es más abundante en las células hematopoyéticas y epiteliales.
En general, las concentraciones de PGRN en el cerebro son bajas; sin embargo, son abundantes en ciertos grupos celulares, como las neuronas piramidales corticales, las células de Purkinje en el cerebelo, las neuronas piramidales del hipocampo y en la microglía (esto según modelos animales). Se le ha implicado en diferentes procesos, como la cicatrización, la promoción del crecimiento celular, la generación de neoplasias y la inflamación.
En rasgos generales, la PGRN tiene un efecto antiinflamatorio, mientras las granulinas ejercen un efecto opuesto. Actualmente se conoce poco acerca del papel de la PGRN en el sistema nervioso central (SNC). Se sabe que su expresión es alta en el neuroepitelio durante el desarrollo embrionario y restringido a ciertas zonas del SNC, por lo cual se le atribuye una función importante en el neurodesarrollo embrionario. Además, se ha comprobado su importancia, en modelos animales, en la promoción de la diferenciación masculina del hipotálamo. Igualmente, es un promotor del crecimiento celular en líneas neuronales en cultivo, y en modelos animales de trauma craneoencefálico se ha demostrado el aumento de sus cantidades cerebrales 24 horas después del trauma. Esto sugiere un posible papel en la supervivencia neuronal (61-63).
El gen de la PGRN se encuentra a tan sólo 2 Mb del gen de MAPT. Contiene 12 exones (32) y los factores que regulan su expresión son poco conocidos, pero se sabe que presenta secuencias regulatorias potenciales, algunas de las cuales responden a citocinas inflamatorias. Una vez sintetizada, la PGRN puede secretarse de manera intacta, puede ser guardada en vesículas o puede ser procesada para la producción de granulinas (61).
Se han reportado múltiples mutaciones del gen de la PGRN relacionadas con la DFT. Todas estas tienen una característica común: truncan la síntesis de la proteína (32,60,64-68). Dentro de estas mutaciones se encuentran:
• Mutaciones sin sentido (nonsense): la mutación genera un codón de terminación temprano, que trunca la síntesis de la proteína. Ejemplos: Q125X. W386X, R418X, Q468X (32-60).
• Cambio del marco de lectura: la eliminación o inserción de nucleótidos cambia el marco de lectura del gen que genera una proteína anómala o un codón de terminación prematuro. Ejemplos: Q130SfsX124, T382SfsX29 (32-60).
• Mutación en el sitio de corte y empalme 5’ del exón 8: se predice que excluye el exón 8 del ARNm y cambia el marco de lectura que produce un codón de terminación prematuro (32,60). En los pacientes que presentan estas mutaciones se halla muy poco ARNm mutante y no se detectan proteínas de PGRN mutantes, por lo cual se concluye que estos son degradados mediante un mecanismo que tiene como función destruir aquellos ARNm que presentan codones de terminación prematuros (32).
• Mutación que altera la secuencia previa al codón de iniciación (secuencia de Kozac) (c.2T>C, M1?) (60).
• Mutación en el sitio de corte y empalme inicial: mutación (IVS0+5G>C) que genera la no codificación del exón 0 y ausencia de ARNm transcrito. Esto puede ser explicado de dos maneras: análisis in silico muestran que esta mutación disminuye la afinidad del ARNm con la U1 snRNP (complejo proteico encargado del corte y empalme). Por ello es posible que este ARNm permanezca con señales que lo retengan en el núcleo y lo lleven, en últimas, a la degradación. Otra posibilidad es que el gen mutado nunca se trascriba (60).
Todas estas mutaciones, mediante diferentes mecanismos, llevan a que el gen no produzca PGRN, con lo cual los niveles de PGRN se reducen a la mitad (haploinsufi- ciencia). Aún cuando no se tiene certeza de la función que cumple la PGRN en el sistema nervioso, se puede deducir mediante la información expuesta que cumple un papel importante en la supervivencia neuronal, el cual es dosis-dependiente. De esta manera, así como se ha demostrado que el exceso de PGRN promueve la tumorogénesis, su deficiencia llevaría a la muerte neuronal y posterior neurodegeneración (32, 60, 62).
No se tiene claridad acerca del papel de las inclusiones positivas para ubiquitina en la fisiopatología de este tipo de DFT. Se considera que la ubiquitina presente en estas inclusiones oculta una proteína precipitada mucho más importante. Se ha descartado la presencia de PGRN en estas inclusiones. Recientemente se ha encontrado que las inclusiones en las demencias frontotemporales con histopatología DFT-ENM, tanto familiares como esporádicas, con mutaciones o sin estas del gen de la PGRN, son positivas para TDP- 43, una proteína que funciona como reguladora de la trascripción, que es ubicua y en las inclusiones se encuentra fosforilada y ubiquitinada. La manera en que se forman estas inclusiones y la acción patológica que estas ejercen se desconocen hasta el momento (69,70).
Presenilina 1
El gen de la presenilina 1, asociado más frecuentemente a la enfermedad de Alzheimer de inicio temprano, se ha encontrado mutado en una familia con características clínicas e histopatológicas de DFT y en un paciente con diagnóstico clínico de DFT (71).
Apo E2
Varios autores han reportado un posible papel del gen Apo E2 como factor de riesgo para DFT; sin embargo, un metanálisis concluyó que esta aseveración requiere mayor confirmación, debido a la heterogeneidad entre los estudios (72).
DFT-3 y DFT-9
Se encuentra reportada en la literatura médica una gran familia en Dinamarca con DFT autosómicadominante, sin patrón histopatológico distintivo, cuya herencia se encuentra ligada a la región pericentromérica del cromosoma 3. Recientemente se ha encontrado en los miembros afectados de esta familia una mutación en el gen de la proteína cargada del cuerpo multivesicular 2B (CHMP2B), el cual parece ser el gen causal en esta familia. Este hallazgo requiere mayores estudios para confirmar la causalidad y el papel de este gen en otros pacientes con DFT (73,74).
También se han encontrado familias con esclerosis lateral amiotrófica y demencia frontotemporal (ELA-DFT) con herencia ligada al locus 9p, específicamente entre las regiones 9p13.2-21.3. La presencia conjunta de estas dos patologías neurológicas en estas familias corrobora el frecuente solapamiento clínico e histopatológico que hay entre ellas. No se ha hallado el gen causal (75,76).
Tratamiento
Estudios han mostrado que existen trastornos serotoninérgicos en la DFT; por lo tanto, el manejo farmacológico ha estado centrado en aquellos fármacos que tienen efecto sobre este neurotransmisor. Los inhibidores selectivos de la recaptación de serotonina (ISRS), entre ellos la paroxetina, han mostrado, en diversos estudios, una mejoría en los síntomas comportamentales de la FTDc, pero no sobre las deficiencias cognoscitivas (3,26,77,78); sin embargo, un estudio más reciente, doble ciego y aleatorizado, no mostró diferencia entre la paroxetina y el placebo en la mejoría de los síntomas comportamentales (79).
Por otro lado, varios estudios han mostrado los beneficios de la trazodona en el manejo de los síntomas comportamentales (26,78,80). A pesar de esto, es necesario profundizar en este aspecto para confirmar su beneficio (81). Otras investigaciones han mostrado efectos favorables de la rivastigmina y el metilfenidato, los cuales requieren mayor sustento (82,83).
Los pacientes con APP pueden beneficiarse de formas alternativas de comunicación, como el lenguaje de señas, tarjetas laminadas informativas, sintetizadores de voz o computadores personales que guardan palabras y frases (8).
Es igualmente importante la intervención no farmacológica del paciente y, sobre todo, de su cuidador, pues se le debe suministrar información sobre la patología y apoyo ante los profundos cambios familiares y económicos que esta enfermedad acarrea (3,26).
Conclusión
La DFT es una enfermedad cuya etiología está en proceso de esclarecerse, con lo cual las bases genéticas de esta han cobrado mayor importancia. Sabiendo esto, es necesario considerar un abordaje genético en el momento de en frentarse a un paciente con DFT e indagar profundamente en los antecedentes familiares, para lo cual es de gran utilidad la realización de un árbol genealógico que nos permita determinar si nos encontramos ante un caso de DFT familiar y el tipo de herencia que presenta.
Sumado a esto, la aplicación de pruebas moleculares en los casos de DFT familiar nos permitiría determinar la presencia o no del gen mutado y con ello el riesgo de los miembros de la familia afectada de sufrir o transmitir la enfermedad a la siguiente generación. Cabe resaltar, sin embargo, que los genes hasta ahora conocidos no explican todos los casos de DFT familiar. Teniendo toda esta información a la mano es posible realizar una adecuada asesoría genética que permita aclarar las principales dudas que presentan los pacientes y sus familias a este respecto.
Referencias
1. Snowden JS, Neary D, Mann DM. Frontotemporal dementia. Br J Psychiatry 2002;180: 140–3. [ Links ]
2. Graff-Radford N, Woodruff B. Frontotemporal Dementia. Semin Neurol 2007; 27: 48–57. [ Links ]
3. Weder N, Aziz R, Wilkins K, Tampi R. Frontotemporal Dementias: A Review. Annals of General Psychiatry 2007; 6:15. [ Links ]
4. Neary D, Sowden J, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546-1554. [ Links ]
5. McKhann G, Albert M, Grossman M, Miller B, Dickson D, et al. Clinical and Pathological Diagnosis of Frontotemporal Dementia. Report of the Work Group on Frontotemporal Dementia and Pick´s Disease. Arch Neurol. 2001; 58:1803-1809. [ Links ]
6. Greicius M, Geschwind M, Miller B. Presenil dementia syndromes: an update on taxonomy and diagnosis. J Neurol Neurosurg Psychiatry 2002; 72:691-700. [ Links ]
7. Mesulam M. Slowly progressive aphasia without generalized dementia. Annals of Neurology 1982; 11:592-598. [ Links ]
8. Mesulam M. Primary Progressive Aphasia – A Language-Based Dementia. N Engl J Med 2003; 349:1535-42. [ Links ]
9. Westbury C. Primary Progressive Aphasia: A review of 112 cases. Brain and Language 1997; 60:381-406. [ Links ]
10. Kertesz A, Blair M, McMonagle P, Munoz D. The Diagnosis and Course of Frontotemporal Dementia. Alzheimer Dis Assoc Disord 2007; 21: 155-163. [ Links ]
11. Ratnavalli E, Brayne C, Dawson K, Hodges J. The prevalence of frontotemporal dementia. Neurology. 2002; 11: 1615-21. [ Links ]
12. Rosso S, Donker L, Baks T, Joosse M, Koning I, Pijnenburg Y et al. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003; 126: 2016-2022. [ Links ]
13. Munoz D, Dickson D, Bergeron C, Mackenzie I, Delacourte A, Zhukareva V. The Neuropathology and Byochemistry of Frontotemporal Dementia. Annals of Neurology 2003; 54 suppl 5: S24-28. [ Links ]
14. Kertesz A, McMonagle P, Blair M, Davidson W, Munoz D. The evolution and pathology of frontotemporal dementia. Brain 2005; 128: 1996-2005. [ Links ]
15. Beteta E. Neuropatología de las Demencias. Revista de Neuro-Psiquiatría 2004; 67: 80-105. [ Links ]
16. Shi J, Shaw C, Plessis D, Richardson A, Bailey K, Julián C, et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 2005; 110: 501-512. [ Links ]
17. Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, et al. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are inmunopositive for p62. Neuroscience Letters 2003; 342: 41-44. [ Links ]
18. Jackson M, Lennox G, Lowe J. Motor neuron disease-inclusion dementia. Neurodegeneration 1996; 5: 339-50. [ Links ]
19. Mackenzie I, Baborie A, Pickering- Brown S, Du Plessis D, Jaros E, Perry R, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuopathol 2006; 112: 539-549. [ Links ]
20. Kertesz A, Kawarai T, Rogaeva E, St. George P, Poorkaj P, Bird T. Familial frontotemporal dementia with ubiquitin- positive, tau-negative inclusions. Neurology 2000; 54: 818-27. [ Links ]
21. Sampathu D, Neumann M, Kwong L, Chou T, Micsenyi M, Truax A, et al. Pathological Heterogeneity of Frontotemporal Lobar Degeneration with Ubiquitin-Positive Inclusions Delineated by Ubiquitin Inmunohistochemistry and Novel Monoclonal Antibodies. Am J Pathol 2006; 169: 1343-1352. [ Links ]
22. Kertesz A, Martinez P, Davidson W, Munoz D. The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology 2000; 55: 1368-1375. [ Links ]
23. Tan C, Piao Y, Kakita A, Yamada M, Takano H, Tanaka M, et al. Frontotemporal dementia with co-occurrence of astrocytic plaques and tufted astrocytes, and severe degeneration of the cerebral white matter: a variant of corticobasal degeneration? Acta Neuropathol 2005; 109: 329-338. [ Links ]
24. Johnson J, Diehl J, Mendez M, Neuhaus J, Shapira J, Forman M, et al. Forntotemporal Lobar Degeneration. Demographic Characteristics of 353 Patients. Arch Neurol 2005; 62: 925- 930. [ Links ]
25. Miller B, Diehl J, Freedman M, Kertesz A, Mendez M, Rascovsky K. International Approaches to Frontotemporal Dementia Diagnosis: From Social Cognition to Neuropsychology. Annals of Neurology 2003; 54 suppl 5: S7-S10. [ Links ]
26. Pasquier F, Fukui T, Sarazin M, Pijnenburg Y, Diehl J, Grundman M, et al. Laboratory Investigations and Treatment in Frontotemporal Dementia. Annals of Neurology 2003; 54 suppl 5: S32- S35. [ Links ]
27. Doval O, Gaviria M. Demencia Frontotemporal. Una redimensiòn de la enfermedad de Pick. Rev colomb psiquiatr 2000; 29:127-54. [ Links ]
28. Rosso S, Landweer E, Houterman M, Doonker L, Van Duijn C, Van Swieten J. Medical and environmental risk for sporadic frontotemporal dementia: a retrospective case-control study. J Neurol Neurosurg Psychiatry 2003; 74: 1574-6. [ Links ]
29. Bird T, Knopman D, VanSweiten J, Rosso S, Feldman H, Tanabe H, el al. Epidemiology and Genetics of Frontotemporal Dementia/Pick’s Disease. Annals of Neurology 2003; 54 suppl 5: S29-31. [ Links ]
30. Chow T, Miller B, Hayashi V, Geschwind D. Inheritance of Frontotemporal Dementia. Arch Neurol 1999; 56: 817- 822. [ Links ]
31. Hutton M, Lendon C, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5´-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702-5. [ Links ]
32. Baker M, Mackenzie I, Pickering S, Gass J, Rademarkers R, Linholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916-19. [ Links ]
33. Yamaoka L, Welsh K, Hulette C, Gaskell C, Murray M, Rimmler J, et al. Linkage of frontotemporal dementia to chromosome 17: clinical and neuropathological characterization of phenotype. Am J Hum Genet 1996; 59: 1306-1312. [ Links ]
34. Goode B, Chau M, Denis P, Feinstein S. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. J Biol Chem 2000; 275: 38182-38189. [ Links ]
35. Andreadis A. Tau gene alternative splicing: expresión patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochimica et Byophysica Acta 2005; 1739: 91-103. [ Links ]
36. Bird T, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, et al. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L). Brain 1999; 122: 741-756. [ Links ]
37. Tolnay M, Grazia M, Rizzini C, Eccles D, Lowe J, Ellison D. A new case of frontotemporal dementia and parkinsonism resulting from an intron 10 + 3 splice site mutation in the tau gene: clinical and pathological features. Neuropathology and Applied Neurobiology 2000; 26: 368-378. [ Links ]
38. Neumann M, Mittelbronn M, Simon P, Vanmassenhove B, de Silva R, Lee A, et al. A New family with frontotemporal dementia with intronic 10+3 splice site mutation in the tau gene: neuropathology and molecular effects. Neuropathology and Applied Neurobiology 2005; 31: 362-373. [ Links ]
39. Lladò A, Esquerra M, Sànchez R, Rami L, Tolosa E, Molinuevo J. A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. European Journal of Neurology 2007; 14: e9-e10. [ Links ]
40. Slowinski J, dominik J, Uitti R, Ahmed Z, Dickson D, Wszolek Z. Frontotemporal dementia and parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology 2007; 27: 73-80. [ Links ]
41. Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N, et al. Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol 2001; 102: 285-292. [ Links ]
42. Bronner I, Meulen B, Azmani A, Severijnen L, Willemsen R, Kamphorst W, et al. Hederitary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain 2005; 128: 2645-2653. [ Links ]
43. Pickering S, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, et al. Frontotemporal dementia with Picktype histology associated with Q336R mutation in the tau gene. Brain 2004; 127: 1415-1426. [ Links ]
44. Boeve B, Tremont I, Waclawik A, Murrell J, Hermann B, Jack C, et al. Longitudianl characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain 2005; 128: 752-772. [ Links ]
45. Kobayashi K, Hayashi M, Kidani T, Ujike H, Iijima M, Ishihara T, et al. Pick’s disease pathology of a missense mutation of S305N of frontotemporal dementia and parkinsonism linked to chromosome 17: another phenotype of S305N. Dement Geriatr Cogn Disord 2004; 17: 293-7. [ Links ]
46. Van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, van de Graaf R, et al. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 2003; 54: 573-81. [ Links ]
47. Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 1999; 58: 667-77. [ Links ]
48. Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, et al. A novel L266V Mutation of the tau gene causes frontotemporal dementia with unique tau pathology. Ann Neurol 2003; 53: 133-7. [ Links ]
49. Miyamoto K, Kowalska A, Hasegawa M, Tabira T, Takahashi K, Araki W, et al. Familial frontotemporal dementia and parkinsonism with a novel mutation at an intron 10+11-splice site in the tau gene. Ann Neurol 2001; 50: 117-20. [ Links ]
50. Pickering S, Baker M, Yen S, Liu W, Hasegawa M, Cairns N, et al. Pick’s disease is associated with mutations in the tau gene. Ann Neurol 2000; 48: 859-67. [ Links ]
51. Murrell J, Spillantini M, Zolo P, Guazelli M, Smith M, Hasegawa M, et al. Tau gene mutation G389R causes a tauopathy with abundant pick bodylike inclusions and axonal deposits. J NEuropathol Exp Neurol 1999; 58: 1207-26. [ Links ]
52. Delisle M, Murrell J, Richardson R, Trofatter J, Rascol O, Soulages X, et al. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol (Berl) 1999; 98: 62- 77. [ Links ]
53. Wszolek Z, Tsuboi Y, Ghetti B, Pickering S, Baba Y, Cheshire W. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet Journal of Rare Diseases 2006; 1:30. [ Links ]
54. Grundke I, Iqbal K. Tau pathology generated by overexpression of tau. AJP 1999; 155: 1781-85. [ Links ]
55. Avila J, Santa-Marìa I, Pèrez M, Hernàndez F, Moreno F. Tau Phosphorylation, Aggregation, and Cell Toxicity. J Biomed Biotechnol. 2006; 3: 74539. [ Links ]
56. Rosso S, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Neirmeijer M, et al. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain 2001; 124: 1948-1957. [ Links ]
57. Mackenzie I, Baker M, West G, Woulfe J, Qadi N, Gass J, et al. A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 2006; 129: 853-867. [ Links ]
58. Kertesz A, Kawarai T, Rogaeva E, St. George P, Poorkaj P. Bird T, et al. Familial frontotemporal dementia with ubiquitin-positive, tau-negative inclusions. Neurology 2000; 54: 818-27. [ Links ]
59. Mackenzie I, Butland S, Devon R, Dwosh E, Feldman H, Lindholm C, et al. Familial frontotemporal dementia with neuronal intranuclear inclusions is not a polyglutamine expansion disease. BMC Neurology 2006; 6:32. [ Links ]
60. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D. Null mutations in progranulin cause ubiquitin- positive frontotemporal dementia liked to chromosome 17q21. Nature 2006; 442: 920-24. [ Links ]
61. Bateman A, Bennett H. Granulins: the structure and functions of an emerging family of growth factors. Journal of Endocrinology 1998; 158: 145-151. [ Links ]
62. Ahmed Z, Mackenzie I, Hutton M, Dickson D. Progranulin in frontotemporal lobar degeneration and neuroinflamation. Journal of Neuroinflammation 2007; 4: 7. [ Links ]
63. He Z, Bateman A. Progranulin (granulin- epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 2003; 81: 600-612. [ Links ]
64. Bronner I, Rizzu P, Seelaar H, van Mil S, Anar B, Azmani A, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. European Journal of Human Genetics 2007; 15: 369-374. [ Links ]
65. Boeve B, Baker M, Dickson D, Parisi J, Giannini C, Josephs K, et al. Fontotemporal dementia and parkinsonism associated with the IVSI+IG>A mutation in progranulin: a clinicopathologic study. Brain 2006; 129: 3103-3114. [ Links ]
66. Snowden J, Pickering S, Mackenzie I, Richardson A, Varma A, Neary D, et al. Progranulin gene mutations associated frontotemporal dementia and progressive non-fluent aphasia. Brain 2006; 129: 3091-3102. [ Links ]
67. Spina S, Murrell J, Huey E, Wassermann E, Pietrini P, Baraibar M, et al. Clinicopathologic features of frontotemporal dementia with progranulin sequence variation. Neurology 2007; 68: 820-827. [ Links ]
68. Cruts M, Kumar S, Van Broeckhoven C. Progranulin mutations in ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Curr Alzheimer Res 2006; 3: 485-91. [ Links ]
69. Cairns N, Neumann M, Bigio E, Holm I, Troost D, Hatanpaa K, et al. TDP-43 in familiar and sporadic frontotemporal degeneration with ubiquitin inclusions. Am J Pathol 2007; 171: 227-240. [ Links ]
70. Seelar H, Schelhaas H, Azmani A, Küsters B, Rosso S, Majoor D, et al. TDP-43 pathoogy in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain 2007; 130: 1375-85. [ Links ]
71. Méndez M, McMurtray A. Frontotemporal- like phenotypes associated with presenilin-1 mutations. Am J Alzheimers Dis Other Demen 2006; 21: 281. [ Links ]
72. Verpillat P, Camuzat A, Hannequin D, Thomas C, Puel M, Belliard S, et al. Apolipoprotein E gene in frontotemporal dementia: an association studyand meta-analysis. Eur J Hum Genet 2002; 10: 399-405. [ Links ]
73. Gydesen S, Brown J, Brun A, Chakrabarti L, Gade A Johannsen P. Chromosome 3 linked frontotemporal dementia (FTD-3). Neurology 2002; 59: 1585-1594. [ Links ]
74. Skibinski G, Parkinson N, Brown J, Chakrabarti L, Lloyd S, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005; 37: 806-808. [ Links ]
75. Valdmanis P, Dupre N, Bouchard J, Camu W, Salachas F, Meininger V, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementa with evidence of linkage to chromosome 9p. Arch Neurol 2007; 64: 240-245. [ Links ]
76. Momeni P, Schymick J, Jain S, Cookson M, Cairns N, Greggio E, et al. Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurology 2006; 6: 44. [ Links ]
77. Moretti R, Torre P, Antonello R, Cazzato G, Bava A. Frontotemporal dementia: paroxetine as a posible treatment of behavior symptoms. A randomized controlled, open 14-month study. Eur Neurol 2003; 49: 13-9. [ Links ]
78. Swartz J, Miller B, Lesser I, Darby A. Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors. L Clin Psychiatry 1997; 58: 212-6. [ Links ]
79. Deakin J, Rahman S, Nestor P,Hodges J, Sahakian B. Paroxetine does not improve symptoms and impairs cognition in frontotemporal dementia: a double-blind randomized controlled trial. Psychopharmacology 2004; 172: 400-8. [ Links ]
80. Lebert F, Stekke W, Hasenbroekx C, Pasquier F. Frontotemporal dementia: a randomized controlles trial with trazodone. Dement Geriatr Cogn Disord 2004; 17: 355-9. [ Links ]
81. Martinon-Torres G, Fioravanti M, Grimley Evans J. Trazodone for agitation in dementia. Cochrane Database of Systematic Reviews 2004, Issue 3. Art. No.: CD004990. DOI: 10.1002/14651858.CD004990. [ Links ]
82. Rahman S, Robbins T, Hodges J, Mehta, Nestor P, Clark L, et al. Methylphenidate (‘Ritalin’) can ameliorate abnormal risk-taking behaviour in the frontal variant of frontotemporal dementia. Neuropsychopharmacology 2006; 31: 651-8. [ Links ]
83. Moretti R, Torre P, Antonello R, Cattaruzza T, Cazzato G, Bava A. Rivastigmine in frontotemporal dementia: an open label study. Drugs Aging 2004; 21: 931-7. [ Links ]
Recibido para evaluación: 5 de octubre de 2007 Aceptado para publicación: 16 de enero de 2008

