










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.55 no.2 Bogotá Apr./June 2007
PRESENTACIÓN DE CASOS
Yolanda Cifuentes C.1, Martha Bermúdez2 Clara Arteaga D.3
1. Pediatra Neonatóloga, Profesor Asociado División de Neonatología Instituto Materno Infantil, Facultad de Medicina, Universidad Nacional de Colombia.Bogotá.
2. Bióloga, Unidad de Biología de la Procreación, Instituto Materno Infantil, Universidad Javeriana, Universidad Colegio Mayor de Cundinamarca.
3. MSc Genética, Profesor Asociado Departamento de Ginecología y Obstetricia, Coordinador Unidad Biología de la Procreación, Instituto Materno Infantil. Correspondencia: mycifuentesd@unal.edu.co
Resumen
La hiperglicinemia no cetósica (HGNC) es un error innato del metabolismo, de carácter recesivo, debido a un defecto en el sistema de clivaje, que ocasiona acumulación de glicina en la sangre y en el sistema nervioso central, donde activa dos receptores diferentes. El receptor localizado en la médula espinal, inhibitorio, provoca la apnea y el hipo de estos pacientes. El receptor cortical, excitatorio, produce la lesión cerebral y las convulsiones. Se presenta la historia de un recién nacido masculino, a término, que se torna encefalopático en los primeros días de vida. No presentó hipoglicemia, acidosis metabólica, cetosis ni hiperamonemia. La ecografía transfontanelar fue normal. A los seis días ameritó ventilación mecánica. La cromatografía en capa fina mostró banda de glicina en plasma y orina. El paciente recibió manejo con benzoato de sodio, diazepam y restricción proteica. A los 20 días de vida presentó mejoría neurológica y salió del hospital a los 42 días de vida con igual manejo. El estudio de acilcarnitinas en sangre por tandem-masas hecho en Santiago de Compostela fue normal. La cuantificación de aminoácidos hecha en el CEDEM mostró aumento de los niveles de glicina en plasma y LCR y la relación glicina LCR / plasma confirmó el diagnóstico de hiperglicinemia no cetósica típica. Se revisan las causas de encefalopatía neonatal y se plantea una secuencia para el diagnóstico.
Palabras clave: encefalopatías, hiperglicinemia no cetósica (HGNC), errores innatos del metabolismo (EIM), fenotipo.
Cifuentes Y, Bermúdez M, Arteaga C. Encefalopatía neonatal, algo mas que asfixia al nacer. rev.fac.med. 2007; 55: 126-134.
Summary
Nonketotic hyperglycinemia is an inborn error of metabolism resulting from a defect in the glycine cleavage enzyme system, is inherited as an autosomal recessive trait. It is characterized by elevated concentrations of glycine in blood and central nervous system. Accumulation of glycine in the brain is thought to cause excessive stimulation of the inhibitory and excitatory receptors presenting apnea, hipcus, seizures and brain damage. We report a full-term male newborn with encephalopathy in the first days of life. He did not present hypoglicemia, metabolic acidosis, non hyperammonemia cetosis. The brain ultrasonography was normal. At six days of age he presents respiratory failure and needs mecanic ventilation. Thin layer chromatography shows glycine in plasma and urine. He was treated with sodium benzoate, diazepam and a low-protein diet. He improved and at 42 days of age he was discharged with the same treatment. The acylcarnitine profile does in Santiago de Compostela was normal. The concentrations of amino acids does in CEDEM was found elevated glycine in plasma and cerebrospinal fluid and the glycine ratio LCR/plasma establish the diagnosis of Typical Neonatal non-ketotic hyperglycinemia. We revised the etiology of neonatal encephalopathy and proposed diagnostic secuency.
Key words: brain diseases, hyperglycinemia, nonketotic, metabolism, inborn errors (IEM), phenotype.
Cifuentes Y, Bermúdez M, Arteaga C. Neonathal encefhalopathy something more that perinatal asphysia. rev.fac.med. 2007; 55: 126-134.
Introducción
La glicina es un aminoácido no esencial, el más sencillo y abundante. Es catabolizada en distintos tejidos a través de un complejo enzimático mitocondrial.
La hiperglicinemia no cetósica (HGNC) es un error innato del metabolismo de la glicina, de carácter recesivo, debido a un defecto en el sistema de clivaje de la glicina localizado en el interior de la membrana mitocondrial en hígado, riñón, cerebro y placenta. El complejo enzimático está formado por cuatro proteínas: una proteína P dependiente de fosfato de piridoxal; una proteína T dependiente de tetrahidrofolato, una aminometiltransferasa (H) y una deshidrogenasa dependiente de lipoamida (L).
La mayoría de los pacientes con HGNC carece de proteína P, habiéndose mapeado el gen en el cromosoma 9p13 o 9p23-24.
Los defectos en la actividad de este sistema llevan a la acumulación de glicina fundamentalmente en el sistema nervioso central, donde activa dos receptores diferentes. El receptor clásico, localizado en la médula espinal, tiene funciones inhibitorias, lo que provoca la apnea y el hipo de estos pacientes. El receptor en la corteza cerebral, N-metil-D-aspartato (NMDA), tiene funciones excitadoras y su activación produce la lesión cerebral y las convulsiones (1,2).
Existen dos fenotipos: la forma típica en la cual la actividad del sistema de clivaje está ausente o es muy baja y la relación glicina en LCR/plasma está en el rango 0.098-0.9. En la forma atípica de la enfermedad hay actividad residual del sistema de clivaje y la relación LCR/plasma está en el rango de 0.09-0.097 (3).
En la edad pediátrica existen varias formas de presentación clínica: neonatal, transitoria e infantil. En la forma neonatal típica, los pacientes normales al nacer, en los primeros días de vida presentan hipotonía e incapacidad para succionar, letargia profunda que progresa al coma, algunos requieren ventilación mecánica y mueren en las primeras semanas de vida. Los que sobreviven presentan un severo daño neurológico. Muchos pacientes mueren en forma rápida sin que se llegue a sospechar el diagnóstico.
La forma transitoria es clínicamente indistinguible de la forma neonatal, entre las dos y ocho semanas de vida hay retorno a los niveles normales de glicina en LCR y plasma con remisión de las manifestaciones clínicas, el pronóstico neurológico de estos pacientes, en general, es bueno. Cuatro de cinco infantes descritos no tenían alteración neurológica en un seguimiento que varió entre los seis meses y los cuatro años. Un caso tenía severo retardo del desarrollo a la edad de nueve meses (4,5).
En la forma infantil, el crecimiento y desarrollo son normales hasta los seis meses de vida y posteriormente presentan apneas, convulsiones y moderado retardo mental.
Existen formas de presentación tardía. Una de ellas a partir del segundo y tercer año con crisis convulsivas y algún grado de retraso mental. Otra aparece aún en épocas posteriores de la niñez, con diplejía espástica progresiva y atrofia óptica, pero sin afectación cognitiva. Estas formas de presentación tardía aparecen como consecuencia de anomalías en la proteína H o T (2).
El EEG realizado en las dos primeras semanas de vida muestra un patrón característico de estallido-supresión. Este patrón electroencefalográfico cambia y a los tres meses de edad se puede observar un trazado de hipsarritmia.
El patrón de estallido-supresión no es diagnóstico de HGNC pues otras entidades como encefalopatía hipóxico-isquémica, meningitis, encefalopatía epiléptica infantil precoz (síndrome de Ohtahara), epilepsia mioclónica precoz y leucinosis se han informado como causa del patrón estallido-supresión (6). Los pacientes con HGNC pueden presentar mioclonías en asociación con otros tipos de convulsiones y posteriormente desarrollar espasmos infantiles con hipsarritmia (1,7-9).
Los recién nacidos afectados de HGNC pueden tener daño cerebral in útero. Varias publicaciones han informado anomalías estructurales del sistema nervioso como agenesia del cuerpo calloso, ventriculomegalia, hidrocefalia, colpocefalia (11-15). La disgenesia o agenesia de cuerpo calloso también se ha descrito en otros errores innatos del metabolismo como acidosis láctica primaria, síndrome de Zellweger, adrenoleucodistrofia, síndrome de Menkes y aciduria glutárica tipo II. Algunas madres han informado movimientos fetales anormales que podrían corresponder a hipo.
En la RMN se demuestra retardo en la mielinización y disgenesia del cuerpo calloso. La asociación de HNC y hemorragia intracraneal fue descrita recientemente (10).
El diagnóstico se realiza cuantificando la glicina en plasma y LCR, estableciendo la relación entre estas dos determinaciones. La relación LCR/ plasma mayor de 0.08 es diagnóstica. El diagnóstico definitivo se realiza midiendo la actividad del sistema de clivaje de glicina en tejido hepático.
El diagnóstico es de gran utilidad para hacer la correspondiente asesoría genética.
Es posible hacer el diagnóstico prenatal de la entidad. Inicialmente se postuló que la relación de glicina/serina en el líquido amniótico podía ser de utilidad, sin embargo, varios autores informan que esta relación no es confiable. La demostración de la disminución significativa de la actividad del sistema de clivaje de glicina en biopsia de vellosidad corial hace el diagnóstico (16).
Recientemente se informaron de tres casos falsos negativos de diagnóstico prenatal con niños afectados, por lo que si se conoce la mutación se puede optar por diagnóstico de DNA (17).
El pronóstico de los pacientes depende fundamentalmente de la forma de presentación, típica o atípica y se correlaciona con la ausencia del sistema de clivaje o la demostración de algún residuo de actividad del sistema de clivaje. No existe tratamiento específico, la exsanguinotransfusión en período neonatal disminuye los niveles de glicina en plasma. Se ha utilizado la estricnina, un potente antagonista de los receptores de glicina con resultados variables (18,19). El diazepam y otras benzodiazepinas son competidores de los receptores de glicina en el sistema nervioso central y existen informes de su utilización (20,21).
El benzoato de sodio a dosis de hasta 750 mg/kg/día, que se une a la glicina, forma hipurato y se excreta por la orina, ha demostrado ser útil. El dextrometorfán un antagonista del receptor NMDA, a dosis entre 5 y 35 mg/kg día ha mostrado efectos benéficos en algunos pacientes con HGNC (22-24).
Los estudios terapéuticos están encaminados a disminuír las concentraciones de glicina y a bloquear su efecto en el receptor N-metil-D-aspartato.
Informe del caso
Se trata de un neonato masculino, de 3400 g, talla 51 cm, PC 35 cm, PT 32 cm, fruto de la tercera gestación de una madre de 30 años, que presentó amenaza de aborto a los tres meses por infección urinaria, recibía tiroxina desde hacía tres años, tenía títulos positivos para herpes y refirió disminución de los movimientos fetales los días previos al parto. No había antecedente de consanguinidad ni de retardo mental. Ingresó al IMI a los tres días de vida, en mal estado general, ictérico, con patrón respiratorio irregular y pobre respuesta a estímulos. FC 90xm, FR 31xm, TA 69/44 TAM 52 Sat 99%.
Hipoactivo, hiporreactivo, hipotónico, hiporrefléxico. Babinsky negativo, prensión palmar y plantar negativas, no abre los ojos, pupilas mióticas 2 mm, débilmente reactivas. Succión negativo, búsqueda negativo, oculocefalógiros negativos, movimientos mioclónicos multifocales. Madre hijo 0 positivo. Gases arteriales con acidosis respiratoria, pruebas de función renal y hepática normales. Hiperbilirrubinemia indirecta. Citoquímico de LCR normal. La ecografía transfontanelar fue normal. El paciente continúo con compromiso neurológico y a los seis días de vida ameritó ventilación mecánica. Se cuantificó amonio: 144 micromol/L, se inició benzoato de sodio y se realizó exsanguinotransfusión. A los nueve días de vida el estudio metabólico mostró pruebas colorimétricas en orina negativas, lactato 2 mmol/l, la cromatografía de aminoácidos en capa fina mostró banda de glicina en plasma y orina. Se hizo restricción proteica, se adicionó diazepam y reajustó la dosis de benzoato de sodio.
El paciente continúo con severo compromiso neurológico, presentó una infección por estafilococo aureus, que respondió bien al tratamiento antibiótico. Se documentó en una radiografía de toráx una fractura de clavícula la cual se inmovilizó. A los 20 días de vida mostró mejoría neurológica, prensión palmar positiva, movimientos espontáneos en las cuatro extremidades, se logró extubar, a los 26 días de vida el peso fue de 4025 gr y el PC de 36.5 cm. Una cromatografía de aminoácidos de control en plasma y orina realizada a los 42 días de vida mostró leve aumento de la banda de glicina en plasma y orina. Se dio salida con fórmula de benzoato de sodio, diazepam, dextrometorfán y dieta con restricción de proteínas.
La Ig M para herpes I y II fue negativa. El EEG mostró trazado de sueño espontáneo anormal por ausencia de la arquitectura propia de las etapas de sueño por las que trascurre el registro y en su lugar presentaba actividad lenta irregular, durante todo el trazo. La RMN cerebral a los dos meses de edad mostró alteración de la señal del lóbulo frontal derecho con estigmas hemorrágicos y de las regiones periatriales por lesión isquémica.
A los tres y medio meses, presentaba peso 5.9 k, talla 63 cm PC 39.3 cm. Mal sostén cefálico, sonríe, fija la mirada, hipertonía distal.
El estudio de acil carnitinas en sangre por tandem-masas realizado en el Laboratorio de Detección Precoz de Metabolopatías. Complejo Hospitalario Universitario de Santiago de Compostela fue normal.
Los resultados de la cuantificación de aminoácidos en muestras de plasma y LCR tomadas a los cuatro días de vida, realizada en el centro de Biología Molecular de la Universidad Autónoma de Madrid hallaron un aumento en las concentraciones de glicina en plasma y LCR con una relación de glicina LCR/plasma de 0,12, datos confirmatorios de hiperglicinemia no cetósica típica (Tabla 1).

Discusión
El caso se trata, entonces, de un neonato a término masculino que al nacer no presenta alteraciones y posteriormente desarrolla encefalopatía. Las causas que originan una encefalopatía en el período neonatal son múltiples: asfixia perinatal, infecciones, hiperbilirrubinemia, dependencia de piridoxina, hipoglicemia, hemorragia intracraneana, anomalías congénitas y errores innatos del metabolismo (EIM). El interrogatorio a la madre y la realización de una historia clínica dirigida a buscar la causa de la encefalopatía es un instrumento muy valioso. Cuando la encefalopatía se debe a asfixia al nacer, es necesario buscar el compromiso de otros órganos: la cuantificación de enzimas cardíacas, la presencia de oliguria, proteinuria, hematuria, la cuantificación de la creatinina, el aumento importante de células rojas nucleadas, la elevación de las enzimas hepáticas, nos darán una visión clara de la presencia de asfixia y su gravedad, llegando a comprometer el cerebro con una encefalopatía. Este recién nacido no presentó hipoglicemia, acidosis metabólica, alteración de la función renal ni hepática. Los gases sanguíneos mostraron acidosis respiratoria concordantes con su estado encefalopático (25,26).
El análisis del LCR descartó una infección bacteriana y los títulos de IgM para herpes I y II fueron negativos. La ecografía transfontanelar fue normal, no se encontró edema cerebral, hemorragia intracraneana o anomalías mayores del sistema nervioso central (SNC).
Recientemente, el Colegio Americano de Obstetricia y Ginecología y la Academia Americana de Pediatría, concluyeron que menos del 25 por ciento de los neonatos con encefalopatía, tiene evidencia de hipoxia durante el nacimiento y por lo tanto la mayoría de los casos se origina en eventos que ocurren antes de iniciar el trabajo de parto (27).
En la literatura se encuentran informes de niños con alteraciones neurológicas graves, cuya etiología se atribuyó a asfixia al nacer y que posteriormente fueron diagnosticados como un error innato del metabolismo (28,29,).
Los errores innatos del metabolismo en conjunto, no son raros. Un estudio realizado en Canadá mostró una frecuencia de 40 casos por 100.000 nacimientos (30). En el Miami Children Hospital, Amed Soliz informó 14 casos en 55.000 nacimientos para una frecuencia de 1 por 3.900 nacidos vivos. Baldellou en el Hospital Infantil Miguel Servet de Zaragoza informó una frecuencia de uno por cada 1.176 nacidos vivos y uno por cada 274 ingresos hospitalarios (31). En un estudio Alemán sobre 250.000 neonatos, se diagnosticaron 106 pacientes afectados de 23 enfermedades para una frecuencia de 1 por cada 2500 RN (32).
En este paciente, al no encontrar la causa de la encefalopatía se sospechó un error innato del metabolismo por lo que se inició su estudio. Los errores innatos del metabolismo en el período neonatal pasan desapercibidos, confundidos con otras patologías frecuentes como sepsis y asfixia al nacer; cuando la presentación clínica es encefalopatía, ésta con frecuencia es el resultado del efecto tóxico de la acumulación de metabolitos en el sistema nervioso central; la mayoría de ellos cruzan la placenta y son metabolizados por la madre, por eso, los recién nacidos parecen normales al nacer con un período libre de síntomas y luego se van deteriorando, sin que haya una razón aparente y sin respuesta a la terapia sintomática (33).
Si a este paciente no se sospecha un error innato del metabolismo, hubiera fallecido sin diagnóstico como lo informa la literatura. No se demostraron sustancias reductoras ni cuerpos cetónicos en las pruebas colorimétricas. Los niveles de amonio y lactato estaban dentro de los rangos normales para la edad y el paciente no presentaba ni hipoglicemia ni acidosis metabólica por lo que no se sospecharon, acidemias orgánicas o acidemia láctica o defectos del ciclo de la urea.
Este paciente no tenía signos dismórficos, ni compromiso renal o hepático que hicieran sospechar una enfermedad peroxisomal. En la cromatografía (en capa fina) de aminoácidos en sangre y orina se encontró aumento en las bandas de glicina.
La hiperglicinemia es un hallazgo bioquímico que puede ser debido a un defecto primario en el sistema de clivaje o a bloqueo enzimático por metabolitos tóxicos como en las acidurias orgánicas o por drogas específicas como el valproato de sodio (34).
El cuadro era compatible con hiperglicinemia no cetósica. Se asemejaba al de los pacientes con hiperglicinemia no cetósica informados previamente (35).
El EEG de este paciente fue anormal pero no tenía el patrón de salva-supresión que se ha descrito en muchos casos de HGNC, pero que puede estar presente en otras patologías. Este hallazgo electroencefalográfico apoya el diagnóstico de HGNC, pero su ausencia no descarta el diagnóstico (10,36).
El paciente respondió a la terapia instaurada, se logró extubar y se dio de alta de nuestra institución a los 42 días de vida. El perfil de acil carnitinas en sangre, realizado en Santiago de Compostela, fue normal lo que permitió descartar una hiperglicinemia secundaria a acidurias orgánicas.
Posteriormente, se enviaron muestras tomadas al cuarto día de vida al Centro de Biología Molecular de la Universidad Autónoma de Madrid para hacer cuantificación de glicina en plasma y LCR confirmándose el diagnóstico de hiperglicinemia no cetósica típica.
En nuestra institución es el cuarto paciente confirmado en un período de 10 años dentro de un grupo de seis pacientes con cuadro clínico compatible con HGNC. En la figura 1 se presenta una propuesta de secuencia diagnóstica.
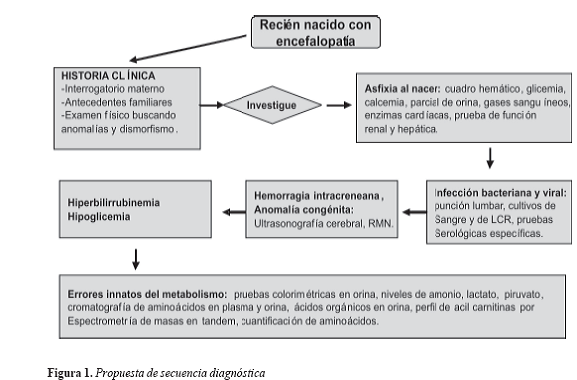
Agradecimiento
A las Doctoras Celia Pérez C. del Centro de Biología Molecular de la Universidad Autónoma de Madrid y Daisy Castiñeiras del Laboratorio de Detección Precoz de Metabolopatías. Complejo Hospitalario Universitario de Santiago de Compostela por la realización de la cuantificación de aminoácidos y el perfil de acilcarnitinas respectivamente.
En homenaje al Instituto Materno Infantil.
Referencias
1. Hamosh A, Johnston MV. Nonketotic hyperglycinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill 8th Ed; 2001: 2065-2078. [ Links ]
2. Casas Fernández C. Aspectos más recientes de la genética de las epilepsias. Rev Neurol. 2000; 30 (supl 1) : S46 - S59. [ Links ]
3. Hayasaka K, Tada K, Fueki N. Nonketotic hyperglycinemia: Analysis of glycine cleavage system in typical and atypical cases. J Pediatr 1987; 110: 873-877. [ Links ]
4. Zammarchi E, Donati MA, Ciani F. Transient Neonatal Nonketotic Hyperglycinemia: A 13-year follow up. Neuroped 1995; 26: 328. [ Links ]
5. Vaquerizo J, Rincón P, Sánchez J, Gómez H, Alejo J, Cardesa JJ. Non-ketotic hyperglycinemia. Transient neonatal form. Rev Neurol 1996; 24: 293-295. [ Links ]
6. Tendero A, López V, Arcas J, Roche MC, Martínez A. Neonatal EEG trace of burst suppression. Etiological and evolutionary factors. Rev Neurol 2001; 33: 514-8. [ Links ]
7. Wang PJ, Lee WT, Hwu WL, Young C, Yau KI, Shen YZ. The controversy regarding diagnostic criteria for early myoclonic encephalopathy. Brain Dev. 1998; 20: 530-5. [ Links ]
8. Bruel H, Boulloche J, Chabrolle JP, Layet V, Poinsot J. Early myoclonic epileptic encephalopathy and non-ketotic hyperglycemia in the same family. Arch Pediatr 1998; 5: 397-9. [ Links ]
9. Chen PT, Young C, Lee WT, Wang PJ, Peng SS, Shen YZ. Early epileptic encephalopathy with suppression burst electroencephalographic pattern-an analysis of eight Taiwanese patients. Brain Dev 2001; 23: 715-20. [ Links ]
10. Lu Fl, Wang PJ, Hwu Wl, Tou Yav KL, Wang TR. Neonatal type of nonketotic hyperglycinemia. Pediatr Neurol. 1999; 20: 295-300. [ Links ]
11. Paupe A, Bidat L, Sonigo P, Lenclen R, Molho M, Ville Y. Prenatal diagnosis of hypoplasia of the corpus callosum in association with non-ketotic hyperglycinemia. Ultrasound in Obstetrics & Gynecology 2002; 20: 616. [ Links ]
12. Mitsui H, Takahashi S, Higano S, Matsumoto K, Shimanuki Y, Ishibashi C, et al. MR imaging findings of nonketotic hyperglycinemia. 2 cases of neonatal onset. Nippon Igaku Hoshasen Gakkai Zasshi 1994; 54: 1047-8. [ Links ]
13. Rogers T, al-Rayess M, O´Shea P, Ambler MW. Dysplasia of the corpus callosum in identical twins with nonketotic hyperglycinemia. Pediatr Pathol 1991; 11: 897-902. [ Links ]
14. Press GA, Barshop BA, Haas RH, Nyhan WL, Glass RF, Hesselink JR. Abnormalities of the brain in nonketotic hyperglycinemia: MR manifestations. Am J Neuroradiol 1989; 10:315-21. [ Links ]
15. Van Hove JL, Kishnani PS, Demaerel P, Kahler SG, Miller C, Jaeken J et al. Acute hydrocephalus in nonketotic hyperglycemia. Neurology 2000; 54: 754-6. [ Links ]
16. Kure S, Rolland MO, Leisti J, Mandel H, Sakata Y, Tada K et al. Prenatal diagnosis of non-ketotic hyperglycinaemia: enzymatic diagnosis in 28 families and DNA diagnosis detecting prevalent Finnish and Israeli-Arab mutations. Prenat Diagn 1999 ; 19: 717-20. [ Links ]
17. Applegarth DA, Toone JR, Rolland MO, Black SH, Yim DK, Bemis G. Non-concordance of CVS and liver glycine cleavage enzyme in three families with non-ketotic hyperglycinaemia (NKH) leading to false negative prenatal diagnoses. Prenat Diagn 2000; 20: 367-70. [ Links ]
18. Arneson D, Chien LT, Chance P, Wilroy RS. Strychnine therapy in nonketotic hyperglycinemia. Pediatrics 1979; 63: 369-373. [ Links ]
19. L von Wendt L, Simila S, Saukkonen Al, Koivisto M. Failure of strychnine treatment during the neonatal period in three Finnish children with nonketotic hyperglycinemia . Pediatrics 1980; 65: 1166-1169. [ Links ]
20. Matalon R, Naidu S, Hughes J, Michals K. Nonketotic hyperglycinemia: treatment with diazepam-a competitor for glycine receptors. Pediatrics 1983; 71: 581. [ Links ]
21. Thio LL, Shanmugam A, Isenberg K, Yamada K. Benzodiazepines Block a2- Containing Inhibitory Glycine Receptors in Embryonic Mouse Hippocampal Neurons. J Neurophysiol 2003 ; 90: 89-99. [ Links ]
22. Alemzadeh R, Gammeltoft K, Matteson K. Efficacy of low-dose dextromethorphan in the treatment of nonketotic hyperglycinemia. Pediatrics 1980; 65: 1166-1169. [ Links ]
23. Hamosh A, Maher JF, Belhs GA, Rasmussen SA, Hohston MV. Long term use of high-dose benzoate and dextromethorphan for the treatment of nonketotic hyperglycinemia. J Pediatr 1998; 132: 709-713. [ Links ]
24. Alemzadeh R, Gammeltoft K, Matteson K. Efficacy of Low-dose dextromethorphan in the treatment of nonketotic hyperglycinemia. Ped 1996; 97: 924. [ Links ]
25. Martin-Ancel A, Garcia-Alix A, Gaya F, Cabanas F, Burgueros M, Quero J. Multiple organ involvement in perinatal asphyxia. J Pediatr 1995; 127: 786-93. [ Links ]
26. Hankins GD, Koen S, Gei AF, López SM, Van Hook JW, Anderson GD. Neonatal organ system injury in acute birth asphyxia sufficient to result in neonatal encephalopathy. Obstet Gynecol 2002; 99: 688-91. [ Links ]
27. ACOG Press Release, January 31, 2003. [ Links ]
28. Feiden W, Bratzke H, Scharschmidt A. Birth injury or congenital brain damage? A case of apparent birth injury with globoid cell leukodystrophy (Krabbe´s disease). Geburtshilfe Frauenheilkd 1991; 51: 65-6. [ Links ]
29. Willis TA, Davidson J, Gray RG, Poulton K, Ramani P, Whitehouse W. Cytochrome oxidase deficiency presenting as birth asphyxia. Dev Med Child Neurol 2000; 42: 414-7. [ Links ]
30. Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. PEDIATRICS 2000; 105: e10. [ Links ]
31. Baldellou A, López J, Rebage V, Salazar MI. Mesa redonda: Errores Congénitos del Metabolismo de Presentación Precoz. Anales Españoles de Pediatría 1998; 20-3. [ Links ]
32. Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization- Tandem Mass Spectrometry: Results, Outcome, and Implications. PEDIATRICS 2003; 111: 1399-1406. [ Links ]
33. Burton BK. Inborn errors of metabolism in infancy: A guide to diagnosis. Pediatrics 1998; 102 : 69. [ Links ]
34. Ciani F, Pasquini E, Ciardetti A, Donati MA, Zammarchi E. Hyperglycinemia in clinical-laboratory practice Pediatr Med Chir 1997; 19: 109-12. [ Links ]
35. Bermúdez M, Arteaga C, Cifuentes Y, Espinosa E, Uribe A, Barrera LA et al. Hiperglicinemia no cetósica (HGNC) forma típica y atípica. Presentación de casos diagnosticados en Colombia. Pediatría 2001; 36: 123-26. [ Links ]
36. Martínez A, Roche MC, López V, Arcas J, Tendero A. Trazado EEG neonatal de salva-supresión. Factores etiológicos y evolutivos. Rev Neurol 2001; 33: 514. [ Links ]
