










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.60 no.3 Bogotá Sept./Dec. 2012
Presentación de casos
Leucoencefalopatía quística no megalencefálica vs. Encefalopatía hipoxicoisquémica: dificultades en el asesoramiento genético
Non-megalencephalic cystic leukoencephalopathy compared to hypoxic ischemic encephalopathy: difficulties in genetic assessment
Deisy Rincón1, Luz Ángela Moreno2, Yenifeth Bello3, Harvy Velasco4
1 Internado Especial Genética Humana. Facultad de Medicina. Universidad Nacional de Colombia, Bogotá.
2 MD, Radióloga, Profesora Asociada Departamento de Imágenes Diagnósticas. Facultad de Medicina. Universidad Nacional de Colombia, Bogotá.
3 MD, Residente, Imágenes Diagnósticas. Facultad de Medicina. Universidad Nacional de Colombia, Bogotá.
4 MD, MSc, Esp; Profesor Asistente, Maestría de Genética Humana. Facultad de Medicina. Instituto de Genética. Universidad Nacional de Colombia, Bogotá.
Correspondencia: hmvelascop@unal.edu.co
Recibido: 15 de junio 2012 / Aprobado: 15 de septiembre 2012
Resumen
Existen varias enfermedades que alteran la conformación de la mielina. Éstas se clasifican en Leucoencefalopatías primarias y secundarias, algunas de ellas con características clínicas, radiológicas, bioquímicas y moleculares que permiten realizar una aproximación diagnóstica. Pese a ello, muchas de éstas tienen cuadros clínicos similares y ya que no se cuenta con ayudas diagnósticas para todas estas patologías, se hace complejo realizar un diagnóstico diferencial. Se presenta el análisis del asesoramiento genético de una pareja con antecedentes de una hija con cuadro de leucoencefalopatía en quien se sospecha una Leucoencefalopatía Quística No Megalencefalica (Leucoencefalopatía primaria) versus una LeucoencefalopatíaHipóxicoIsquemica (Leucoencefalopatía secundaria), donde no se logró llegar a un diagnóstico confirmado por la ausencia de herramientas diagnósticas, sobretodo de índole molecular. Desde esta perspectiva, el asesoramiento genético en esta paciente se constituye en un gran reto para el grupo multidisciplinario que maneja este tipo de casos.
Palabras clave: Leucoencefalopatías, hipóxiaisquémia encefálica, leucomalaciaperiventricular, asesoría genética (DeSC).
Summary
Several diseases alter myelin formation; they are classified into primary and secondary leukoencephalopathies, some of them having clinical, radiological, biochemical and molecular characteristics enabling a diagnostic approach. In spite of this, making a differential diagnosis becomes complicated as many of them have similar clinical pictures and there is a lack of diagnostic aids for all such pathologies. This article analyses the genetic counselling provided for a pair whose background consisted of a daughter having a picture of leukoencephalopathy in whom non-megalencephalic cystic leukoencephalopathy (primary leukoencephalopathy) was suspected as opposed to hypoxic ischemic leukoencephalopathy (secondary leukoencephalopathy), as a confirmed diagnosis could not be reached due to a lack of diagnostic tools, particularly molecular ones. Genetic counselling for this patient thus became a great challenge for the multidisciplinary group managing this type of case.
Keywords: Leukoencephalopathies, Hypoxic-Ischemic Brain, Leukomalacia Periventricular, genetic counseling (MeSH).
Introducción
Existen varias noxas que ocasionan alteraciones en la conformación de la mielina. Estas pueden ser Primarias (Leucodistrofias), donde se afectara principalmente la sustancia blanca y Secundarias donde se afecta principalmente la sustancia gris y otras estructuras cerebrales como ganglios basales y cerebelo. Dentro de las Leucoencefalopatías secundarias se encuentran los desordenes metabólicos, mitocondriales, vasculares, infecciosos y también se incluye la encefalopatía hipóxicoisquémica (1).
La Encefalopatía Hipoxico-Isquémica (EHI) es el síndrome producido por la disminución del aporte de oxígeno (O2) o la reducción mantenida del flujo sanguíneo cerebral (FSC) al encéfalo. Puede ser provocada por una hipoxemia sistémica (asfixia, insuficiencia respiratoria), una alteración en el transporte del O2 (anemia aguda, intoxicación por monóxido de carbono) o una reducción del FSC global (paro cardíaco) (2).
El daño celular en la EHI es multifactorial y su intensidad dependerá de factores como: la edad gestacional del paciente, el grado de hipoxia e hipoperfusión, la glicemia, el pH sanguíneo y la duración de la noxa. Durante períodos de isquemia menores a 5 minutos, mueren sólo neuronas susceptibles del hipocampo y células de Purkinje en el cerebelo. Ante una noxa más prolongada, se pueden producir distintos tipos de compromiso neuropatológico como la necrosis de la capa media de la corteza cerebral, del núcleo amigdalino y de los ganglios basales (3,4).
Por otro lado, las Leucodistrofias son desordenes hereditarios que afectan la sustancia blanca cerebral, presentando alteraciones en la conformación de la mielina. Estas anormalidades primarias de la mielina se agrupan en tres subtipos: los desórdenes Desmielinizantes, los Hipomielinizantes y los Vacuolizantes (1).
Los desórdenes Desmielinizantes se caracterizan por formación anormal de la sustancia blanca o pérdida de la mielina normalmente formada, con preservación de los axones; ejemplos de estos son la adrenoleucodistrofia, la leucodistrofiametacromatica y la enfermedad de krabbe (1).
Las leucodistrofias Hipomielinizantes se caracterizan por disminución de la producción de mielina debido a falla en la síntesis de una de las proteínas específicas de la mielina, por una alteración en el suministro adecuado de precursores de la mielina o por la acumulación de sustancias tóxicas para la oligodendroglía; entre este grupo se encuentran el síndrome de Pelizaeus-Merzbacher, la enfermedad de Alexander, la Hipomielinización con atrofia de los ganglios basales y cerebelo, entre otras (1).
Por otro lado, leucodistrofiasVacuolizantes presentan formaciones quísticas secundarias a la degeneración de la sustancia blanca, en este grupo se encuentran la enfermedad de canavan, la leucoencefalopatíamegaloencefálica con quistes subcorticales (Síndrome de Van der Knaap), leucoencefalopatía con sustancia blanca evanescente(1) y en principio la leucoencefalopatía quística no megaloencefálica (LQNM) (5), aunque nueva evidencia, pone en controversia esta última subclasificación (6).
En este reporte de caso, se presenta el análisis del asesoramiento genético de una pareja con antecedentes de una hija con cuadro de encefalopatía de aparición neonatal, de etiología a establecer.
Reporte de caso
Pareja no consanguínea, G2A0C1V1, mujer de de 24 años de edad y hombre de 30 años de edad, procedentes de la misma región, asisten a genética para asesoría preconcepcional por antecedente de hija con "leucodistrofia", la cual fallece a los 3 meses de vida, secundaria a varias complicaciones.
Presentaron fruto de segunda gestación, femenino, con controles prenatales adecuados, movimientos fetales presentes, TORCH (-), Serología negativa, no exposición a teratógenos; antecedente de placenta previa, polihidramnios y sospecha de atresia esofágica por ecografía; cesárea a las 36 semanas de gestación por abruptioplacentae. Peso al nacer de la última gestación, de 2400 gramos (P10-50), talla 48 cm (P 50-90), perímetro cefálico: 33,6 cm (50-90). Hipotónica, con APGAR 7-9, presentó trastorno de la adaptación requiriendo 2 ciclos de ventilación con presión positiva, con gases de cordón sin trastorno ácido base. Se descarta atresia esofágica.
En las primeras 48 horas de vida presentó dificultad respiratoria, fue trasladada a UCIN y se procedió a intubación oro traqueal. Adicional a esto en el mismo período la paciente presentó crisis convulsivas con clonus - mioclonias en hemicuerpo derecho de difícil manejo y yuguladas hasta el día 7 de vida. Durante los siguientes 10 días se realizaron varias extubaciones fallidas y presentó 2 episodios de paros respiratorios controlados, no se cuantificó el tiempo de apnea. Como comorbilidades se presentó neumonías a repetición y trastorno de la deglución que requirió manejo con gastrostomía.
Al examen físico, se observó piel gruesa, orejas rotadas hacia atrás, facies miopática, ptosis palpebral, micrognatia, estridor inspiratorio y espiratorio, miembros inferiores en hiperextensión que no permitían la flexión pasiva (Figura 1). Valoraciones posteriores de Neuropediatría y Genética plantearon inicialmente un error innato del metabolismo.
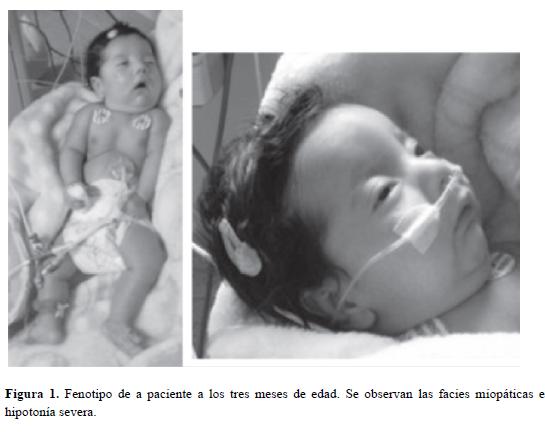
Al mes y 15 días de edad, se reportó microcefalia con cabalgamiento de suturas. Se dió salida a los tres meses con diagnóstico presuntivo (apoyado por RNM) de Síndrome de Van der Knaap y reingresó nuevamente a los dos días por neumonía. Es dada de alta a los tres días, reingresa a los 9 días por dificultad respiratoria. Fallece a los tres meses 15 días de edad.
Dentro de los paraclínicos se encontró: RM cerebral a los 12 días de vida encontrando alteración de la sustancia blanca por lo cual sospechan leucodistrofia.
Gases de cordón antes de las primeras 24 horas: pH 7,289, PCO2 46,8, PO2 10 mmHg, HCO3 22,5, BE -4,5 BEecf: 4,3. Cuadro hemático al nacimiento. Leucocitos 21400, Glóbulos rojos 5.180.000; Hb 18,8; Hcto 54,5, Eosinofilos 2,1%, Plaquetas 165; Neutrofilos 76%.
Ecografía transfontanelar a los 5 días de vida. Sin alteraciones.
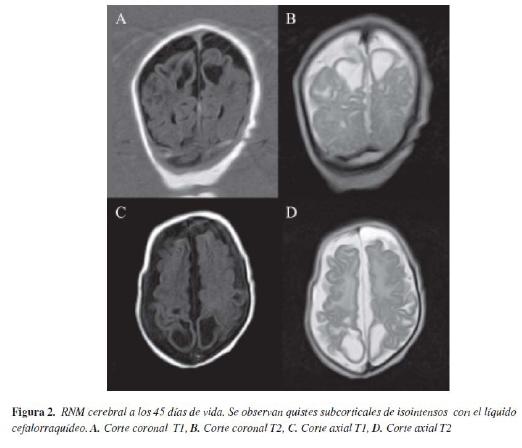
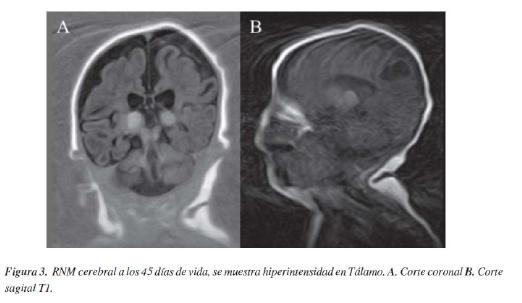
RM cerebral #1 a los 13 días de vida: severa y difusa alteración de la sustancia blanca interpretada por neuropediatría. RM cerebral #2a los 45 días (Figura 2 y 3): extensas zonas de cavitación de sustancia blanca subcortical en ambos hemisferios cerebrales, de predominio parietal, aumento de la intensidad de señal de ambos talamos en la secuencia T1, atrofia cerebral, paquigiria, aumento de tamaño de los ventrículos.
Ácidos grasos de cadena muy larga Normales. Hexosaminidasa Total 792, 72 (control 370,12 nmol/h/mg), Hexosaminidasa A 30,53% control 61%, Hexosaminidasa B 550,45 control 143.82 nmol/h/mg, Arilsulfatasa A: 39,96 (25-80 nmol/h/mg), Iduronato-2-sulfato-sulfatasa 344,4 (140,5nmol/4h/mg), galactosamina 6 sulfato sulfatasa 8,09 (3,21nmol/17h/mg).
De acuerdo a la anterior información, se planteó un diagnostico diferencial conjunto de LQNM vs. una Encefalopatía hipóxico isquémica.
Discusión
En este reporte de caso se muestra la presencia de dos probables diagnósticos diferenciales, uno de origen genético, la LQNM y el otro de origen ambiental, la EHI. Este caso clínico genera un reto muy importante al proceso de asesoramiento genético desde el punto de vista del riesgo de recurrencia, dado los múltiples factores de confusión y de confluencia entre estos dos diagnósticos diferenciales. El abordaje diagnóstico de las leucoencefalopatías es complejo y se inicia empleando datos como los de la anamnesis, antecedentes del paciente y con el apoyo de herramientas como la RNM cerebral. La confirmación de un diagnóstico específico en este grupo de patologías requiere además de análisis bioquímicos, neurofuncionales, moleculares y en muchos de en estos casos, el diagnóstico termina siendo idiopático (Tabla 1)(1,7).
La leucoencefalopatía quística no megaloencefálica, uno de nuestros diagnósticos diferenciales, se caracteriza por presentar aparición infantil, con evolución estable o lentamente progresiva, retraso en el desarrollo psicomotor (el área del lenguaje se puede encontrar muy comprometida y a nivel motor la mayoría de los pacientes logran marcha con apoyo), micro o normocefalia, epilepsia de aparición entre los tres meses a cinco años, espasticidad en el transcurso de la enfermedad y otros síntomas neurológicos como la ataxia, los movimientos atetósicos, alteraciones en la audición, nistagmus, alteraciones en el comportamiento con agresividad y rasgos autistas. También pueden presentar dismorfismo facial con facies de muñeca u otros hallazgos (frente ancha, pliegues epicanticos, hipertelorismo y alteraciones del puente nasal) (5,8). Desafortunadamente es una patología que carece de confirmación molecular de aplicación clínica.
En la RNM cerebral la LQNM, presenta lesiones en sustancia blanca supratentorial que involucra principalmente la región periventricular con conservación de la sustancia blanca central y cerebelar. Las lesiones quísticas subcorticales se observan en la región anterotemporal, con intensidad similar a la del LCR y se puede observar mediante secuencias FLAIR, signos de mielinización anormal periquistica. Otros hallazgos que se pueden encontrar en la RM son aumento del tamaño del cuerno temporal, ventriculomegalia, disminución del volumen cerebral, cavum septum pellucidum, cavumvergae(7,8).
La LQNM es causada por mutaciones homocigotas en el gen RNASET2 (8). Este gen, localizado en 6q27, codifica para la Ribonucleasa T2, del tipo de ribonucleasas extracelulares que pertenece a la familia de RNasas Rh/T2/S, asociándose con malignidad en humanos (9). Esta familia de RNasas T2, son las únicas Rnasas activas en un pH ácido y por consiguiente el RNASET2 se describió como una hidrolasa lisosomal putativa. Recientemente se ha descrito un modelo animal en zebrafish, donde se ha propuesto que la LQNM se comportaría mas como una enfermedad de depósito lisosomal, donde la acumulación de RNA produciría la degeneración axonal y las alteraciones en sustancia blanca (6).
En el caso de nuestro paciente, a pesar de presentar retraso global del desarrollo, microcefalia y hallazgos en RM cerebral de quistes en región anterotemporal, estos hallazgos clínicos no son contundentes a la hora de definir este diagnóstico debido a varios factores como: que los quistes en nuestro paciente son también parietales, probablemente no congénitos, según lo reporta la primera RNM cerebral, la presencia de hiperintensidades en hipotálamo, la aparición temprana de epilepsia y la ausencia de fenotipo de cara de muñeca, de modo que la enfermedad hipóxico isquémica por noxa perinatal (abruptioplacentae y otros antecedentes como dos paros respiratorio), tendría un papel relevante como diagnóstico diferencial.
Esta EHI puede presentar varias manifestaciones clínicas que dependerán del grado de afectación cerebral; en los recién nacidos (RN) con formas leves se observa alteraciones en el tono muscular (hipo/hipertonia), temblores y reflejos primitivos exaltados, en las formas moderadas los RN están letárgicos, hipotónicos y con reflejos débiles, pudiendo aparecer o no convulsiones, mientras que en los RN con EHI grave presentan estupor o coma, convulsiones y ausencia de reflejos primitivos, siendo frecuente la aparición de microcefalia, cuadriplejía espástica y convulsiones de difícil manejo.
La leucomalacia quística se presenta cuando la injuria anóxica es severa y prolongada (dura más de 30 minutos), produciendo un daño difuso en el tejido cerebral (10) donde las neuronas no recuperan su función, desarrollándose posteriormente un infarto en esa región(11). Durante la reperfusión se produce un daño bioquímico causado por los radicales libres de oxígeno y las fosfolipasas activadas por el calcio. Si los amortiguadores de radicales libres (superoxidodismutasa, catalasa, glutationperoxidasa, vitaminas C y E) se ven desbordados, éstos se combinarán con los ácidos grasos insaturados originando peróxidos lipídicos que provocarán reacciones de peroxidación extremadamente tóxicas para las membranas y el DNA celular causando finalmente la muerte neuronal (12).
En las imágenes típicas de encefalopatía hipoxico isquémica, podemos ver el edema cerebral difuso, con hipointensidades en T1 e hiperintensidades en T2, que se detecta en los primeros dos o tres días posterior a la lesión, pero más adelante se podrá observar hiperintensidades marcadas en T1 e hipointensidades en T2 con compromiso de los ganglios basales y del tálamo. Estas lesiones rápidamente se convierten en cavitaciones, respetando el bulbo raquídeo y el cerebelo, dando el aspecto final de leucomalaciamultiquistica (10).
En este caso sería fundamental la utilización de herramientas más allá de la clínica para definir el diagnóstico y por ende el riesgo de recurrencia, el cual es uno de los productos más importantes del proceso de asesoramiento genético. Con el diagnóstico concreto, el genetista, apoyado por el grupo multidisciplinario de trabajadores de la salud puede: definir el porcentaje de riesgo de ocurrencia de un nuevo caso en la familia, establecer el pronóstico y definir las medidas de soporte preventivo y paliativo.
Con el paciente arriba descrito, este proceso de asesoramiento genético podría quedar inconcluso debido a la ausencia de pruebas válidas de uso clínico para descartar la LQNM y se estaría flanqueando de forma teórica el riesgo de recurrencia de un 25%, para la LQNM a un valor menor del 1% en caso de la leucomalacia secundaria a hipoxia. Finalmente el diagnóstico acá será presuntivo, apoyados más por los antecedentes, la clínica y los paraclínicos, hacia una EHI que hacia la LQNM.
Conclusiones
A pesar del vasto conocimiento que se tiene sobre muchas de las características clínicas y paraclínicas de las enfermedades que ocasionan alteraciones en la conformación de la mielina, aun existen vacíos que están por abordar, sobretodo en casos donde se sobrelapan signos y síntomas de las leucoencefalopatías primarias y secundarias.
En los últimos años se han empleado herramientas bioquímicas y moleculares para permitir una mayor diferenciación entre estos dos tipos de leucoencefalopatías, de modo que se pueda llegar a un diagnóstico concreto que a la larga le permita definir al clínico su etiología, pronóstico y riesgo de recurrencia.
Sin embargo, existen aun varias patologías de la mielina por describir, y las recientemente descritas, presentan etiología idiopática o los genes responsables de las mismas no son susceptibles a un análisis paraclínico válido, lo que trae consigo grandes retos en el proceso del asesoramiento genético, el cual no podrá ser el más adecuado en estos casos.
Se requiere de un mayor número de casos descritos de estas patologías raras, además de la posibilidad de disponibilidad de test diagnósticos específicos (bioquímicos, neurofuncionales o moleculares) en las mismas, que permitan, en casos confusos como estos, la ponderación y confirmación o descarte de los diagnósticos diferenciales.
Referencias
1. Lyon G, Fattal-Valevski A, Kolodny EH. Leukodystrophies: Clinical and Genetic Aspects. Top MagnReson Imaging. 2006; 17:219-42. [ Links ]
2. Koons A, Mojica N, Jadeja N, Osffeld B, Hiatt M, Hegyi T. Neurodevelopmental outcome of infants with apnea of infancy. Am J Perinato.1993; 10:208-11. [ Links ]
3. Armstrong-Wells J, Bernard Ti, Boada R, Manco-Johnson M. Neurocognitive outcomes following neonatal encephalopathy. Neuro Rehabilitation.2010; 26: 27-33. [ Links ]
4. Back S. Perinatal White Matter Injury: The changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard DevDisabil Res Rev. 2006; 12:129-140. [ Links ]
5. Henneke M, Preuss N, Engelbrecht V, Aksu F, Bertini E, Bibat G, et ál. Cystic leukoencephalopathy without megalencephaly: A distinct disease entity in 15 children. Neurology.2005; 64:1411-6. [ Links ]
6. Haud N, Kara F, Diekmann S, Henneke M Willer JR, Hillwig MS, et ál. RNASET2 mutant zebrafish model familial cystic leukoencephalopathy and reveal a role for RNase T2 in degrading ribosomal RNA. PNAS.2011; 108:1099-103. [ Links ]
7. Kohlschütter A, Eichler F. Childhood leukodystrophies: a clinical perspective. Expert Rev Neurother. 2011; 11:1485-96. [ Links ]
8. Henneke M, Diekmann S, Ohlenbusch A, Kaiser J. Engelbrecht V, Kohlschütter A, et ál. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet. 2009; 41:773-5. [ Links ]
9. Luhtala N, Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem Sci. 2010; 35:253-9. [ Links ]
10. Triulzi F, Baldoli C, Righini A. Neonatal Hypoxic-lschemic Encephalopathy. En: Tortori-Donati P and Rossi A. Pediatric Neuroradiology: Brain, Head and Neck spine. Berlín: Heidelberg; 2005. [ Links ]
11. Squier W, Austin T, Anslow P Weller R. Infant subcortical cystic leucomalacia: A distinct pathological entity resulting from impaired fluid handling. Early Hum Dev. 2011; 87:421-6. [ Links ]
12. Kellmer I, Hagberg H. Perinatal brain damage, excitatory amino acids and oxygen derived free radicals. En: Van Geijin HP, Copray FJ: A Critical appraisal of fetal surveillance. Amsterdam. Elsevier Science; 1994. [ Links ]
