










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.62 no.2 Bogotá Apr./June 2014
https://doi.org/10.15446/revfacmed.v62n2.45419
http://dx.doi.org/10.15446/revfacmed.v62n2.45419
Casos clínicos
Primer caso reportado en Colombia de hipoplasia renal congénita bilateral en dos hermanos
The first case to be reported in Colombia of bilateral congenital renal hypoplasia in two brothers
José Augusto Urrego-Díaz1; José David Romero-Rueda1; Guillermo Landinez-Millán2; Carlos Javier Lozano-Triana2; Luz Ángela Moreno-Gómez3
1 Facultad de Medicina. Universidad Nacional de Colombia. Bogotá, Colombia.
2 Departamento de Pediatría. Facultad de Medicina. Universidad Nacional de Colombia. Bogotá, Colombia.
3 Departamento de Radiología e Imágenes Diagnósticas. Facultad de Medicina. Universidad Nacional de Colombia. Bogotá, Colombia.
Correspondencia: José Augusto Urrego Díaz. Dirección: Carrera 14B No. 163-29 Apartamento 805. Bogotá, Colombia. Teléfono: +57 18103204.
Correo electrónico: joaurregodi@unal.edu.co
Urrego-Díaz JA, Romero-Rueda JD, Landinez-Millan G, Lozano-Triana CJ, Moreno-Gomez LA. Primer caso reportado en Colombia de hipoplasia renal congenita bilateral en dos hermanos. rev.fac.med. 2014;62:279-285.
Recibido: 12/12/2013 / Aceptado: 29/05/2014
Resumen
Las anormalidades congénitas del riñón y de vías urinarias son la primera causa de falla renal en la niñez. Una de estas anormalidades es la hipoplasia renal, definida como un riñón de tamaño disminuido que conserva su forma y parte de su función. Su presentación unilateral es más común que la bilateral con una relación de 7:1, de tal forma que esta última es una presentación no usual. Así mismo, es escasa la información sobre casos de hipoplasia renal congénita bilateral (HRCB) en miembros de una misma familia. En este artículo se reporta el caso de dos hermanos con HRCB, diagnosticada por ecografía renal y de vías urinarias. El primero de ellos recibió el diagnóstico de HRCB a los cuatro años de edad, tras requerir estudio ecográfico por persistencia de talla baja para la edad en sus controles de crecimiento y desarrollo; el siguiente paciente tenía ocho años de edad al momento del diagnóstico, es hermano del anterior caso y su diagnóstico de HRCB fue hallado en un estudio ecográfico realizado por compromiso en su función renal. En ambos casos el estudio imagenológico reportó disminución en el tamaño renal bilateral según el índice de Hodson. El objetivo principal de este documento es el de dar a conocer esta patología que, si bien es rara, no deja de ser importante su presentación bilateral y su asociación familiar, de las que se tiene poca información en la literatura médica. Se espera que en el futuro esta información sirva para el tamizaje familiar de los pacientes con HRCB.
Palabras clave: Anomalías Congénitas, Riñón, Pediatría, / ultrasonografía (DeCS).
Summary
Congenital anomalies concerning the kidneys and urinary tract represent a heterogeneous group of defects and the main cause of renal failure during childhood. Kidney hypoplasia is one such defect; it is defined in clinical practice as a kidney which is significantly shorter than normal but retains a normal shape and some of its function. Bilateral congenital kidney hypoplasia (BCKH) is 7 times less more frequent than unilateral hypoplasia, thus being one of the rarest anomalies of the kidney and urinary tract. The presence of BCKH in more than one family member is even rarer. This article deals with two male brothers having BCKH diagnosed through ultrasonography of the kidneys and urinary tract. A postnatal diagnosis was made for both of them; the younger brother received his diagnosis when he was 4 years old after being studied for the persistence of low height for his age and the older brother received his at 8 years of age after decreased renal function was revealed during hospitalisation. Ultrasonography study showed reduced size for both kidneys in both cases, according to Hodson's index. This article discusses aspects concerning this congenital anomaly and highlights this disorder's importance in some clinical scenarios. It is hoped that this information may be of use during the initial approach to the family of a BCRH patient.
Key words: Congenital Abnormalities, Kidney, Pediatrics, / ultrasonography (MeSH).
Introducción
Las Anormalidades Congénitas del Riñón y de Vías Urinarias (ACRVU), como vejiga neurogénica, reflujo vesicoureteral, uropatía obstructiva, megaureteres, riñón ectópico, obstrucción de la unión ureteropélvica, displasia renal e hipoplasia renal (1,2), ocurren en aproximadamente el 0,3-0,7% de todos los embarazos (3-5), con una incidencia de dos a tres veces mayor en fetos masculinos que en los femeninos (1,6) y corresponden a un 23% de todos los defectos congénitos evidenciados en recién nacidos (7). Estas anomalías son causa de Enfermedad Renal Crónica (ERC), contribuyen al 40-60% de esta en la población pediátrica a nivel mundial (6,8) y se considera la mayor causa de falla renal en la niñez (9).
Aunque existen reportes de casos, la ocurrencia familiar de algunas de estas anormalidades es sumamente rara (1013), como es el caso de la Hipoplasia Renal Congénita (HRC), que se presenta en el 0,027% de los embarazos (14). Adicionalmente, los casos de HRC bilateral son 7 veces menos frecuentes que aquellos donde hay afección de un solo riñón (15), siendo una de las ACRVU menos frecuentes (6). En este artículo se presenta el caso de dos hermanos con HRC bilateral y se discute esta patología. Es el primer caso reportado en Colombia de HRC bilateral en hermanos.
Caso 1
Paciente masculino de cuatro años de edad, estudiado inicialmente por el servicio de endocrinología pediátrica por síndrome de talla baja (talla de 91 centímetros), que corresponde a tres desviaciones estándar por debajo de la esperada para su edad. Dentro de los resultados de las pruebas de laboratorio solicitadas por esta especialidad se encuentran signos de compromiso renal crónico como: nitrógeno ureico en sangre (BUN) (33,5), creatinina (1,15), Depuración creatinina (45 mg/min/M2), calcio (9,6), fosforo (4,9), PTH (67,6), parcial de orina con pH de 6,5, densidad 1010, proteínas 25 mg/dl, gases arteriales con pH de 7,37 y Hco3 de 20,2 y electrolitos (Na: 141; K: 4.1; Cl: 106).
También se realizó un estudio ecográfico que muestra un riñón derecho 46x29x25 mm e izquierdo con 51x28x28 mm, con un índice de Hodson para el paciente de 8,1; por lo tanto, se informa que el tamaño de los riñones está disminuido de forma bilateral y se diagnostica HRC bilateral. El paciente es remitido al servicio de nefrología pediátrica.
Durante un periodo de seis años el paciente es controlado por este servicio, diagnosticándose insuficiencia renal crónica estadio III por una TFG de 41,9 ml/min/1,73 m2. A pesar del manejo instaurado (con dieta hiposódica e hipoproteica, calcitriol 0,5 mg/ día, enalapril en dosis progresiva según cifras de TA hasta 5 mg dosis única diaria, ácido fólico 1 mg/día, losartán 25 mg/día y carbonato de calcio 600 mg/día), la función renal siguió comprometiéndose de forma progresiva. En su último control (agosto de 2013) el paciente se encuentra poliúrico, con hiperazoemia (Creatinina de 5,4 y BUN de 54), con una relación BUN-creatinina de 10, orina hipoosmolar y un índice de Schwartz de 28 (Estadio IV de insuficiencia renal crónica). El último estudio ecográfico revela pérdida de la relación corticomedular, incremento difuso de la ecogenicidad y persistencia de disminución del tamaño renal, sin cambio alguno en sus dimensiones (longitud riñón derecho 4,6 cm y longitud riñón izquierdo 5,1 cm, como se ve en las figuras 1 y 2, respectivamente). Con estos datos se decide clasificar al paciente como candidato de primera línea para trasplante renal y continuar hasta entonces con el tratamiento instaurado de nefroprotección.
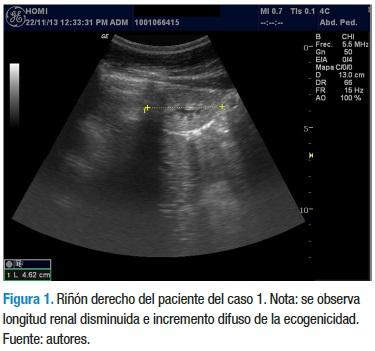
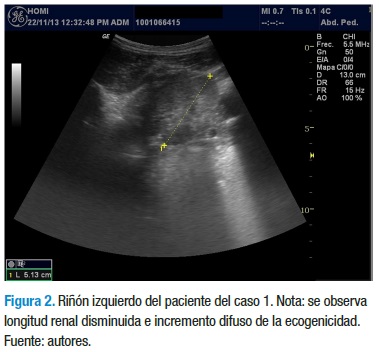
Caso 2
Paciente masculino de ocho años de edad, hermano del caso anterior, quien asiste en compañía de su madre a la consulta externa de nefrología pediátrica; remitido de otra institución en donde se encontró alteración en pruebas de función renal, como: creatinina 0,82, Tasa de Filtración Glomerular (TFG) 76,9 ml/min, BUN 18,5, uroanálisis pH 5,0, densidad 1020. Se interpreta como insuficiencia renal crónica estadio I. Se realiza estudio ecográfico renal y de vías urinarias que muestran un riñón derecho con dimensiones 43X27X23 mm, riñón izquierdo con dimensiones de 67X27X22 mm, para un índice de Hodson de 9,3; se demuestra la hipoplasia renal bilateral.
Con los datos anteriores, el servicio de nefrología pediátrica inicia tratamiento con enalapril 5 mg/día, calcitriol 0,25 mg/ día y dieta rica en carbohidratos, con restricción proteica, de sodio y lácteos. La evolución clínica y paraclínica ha sido estacionaria y sin signos de aumento en el deterioro de la funcional renal, a diferencia de su hermano. En el momento tiene 12 años, asiste al colegio y cursa 5° grado con un buen desempeño escolar. En la última ecografía se reporta una longitud renal de 4,3 cm para el riñón derecho y 6,7 cm para el riñón izquierdo, con aumento de la ecogenicidad en ambos riñones, con menor compromiso del riñón izquierdo (Figuras 3 y 4).
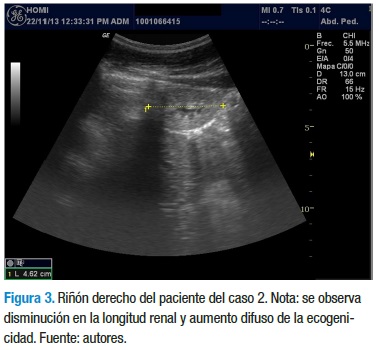
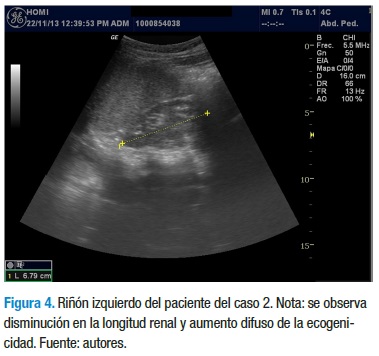
Finalmente, a la madre de estos niños se le realizó un estudió ecográfico para evaluar el tamaño de sus riñones y no se encontró alteraciones. No fue posible realizar el mismo estudio a otros familiares.
Discusión
El diagnóstico de HRC es histológico y se hace al encontrar un riñón con un importante déficit de nefronas secundario a un defecto congénito que no le permite al riñón alcanzar su volumen normal. Entre sus diagnósticos diferenciales está la atrofia renal, en la que el déficit de nefronas es secundario a procesos inflamatorios que afectan el riñón ya formado, como infecciones, glomerulonefritis, entre otros, eventos que no fueron hallados entre los antecedentes de los dos casos presentados. Otro de los diagnósticos diferenciales de la HRC es la displasia renal, que puede cursar también con un tamaño renal disminuido y cuya diferenciación requiere así mismo de un diagnóstico histológico (16). Sin embargo, en muchos casos no es ético realizar biopsias renales en los pacientes, como en los casos aquí reportados, por lo que en la práctica clínica se acepta el uso del término hipoplasia renal cuando el tamaño del riñón es significativamente menor, pero se conserva su forma y parte de su función (17).
Para determinar el tamaño renal en los casos reportados se usó la ecografía renal y de vías urinarias. Existe suficiente evidencia que demuestra cómo el volumen renal medido por este método imagenológico es un indicador bastante exacto del tamaño real del riñón (18,19). Adicionalmente, si bien otros métodos pudieran llegar a ser más exactos, su bajo costo, su seguridad y su naturaleza no invasiva convierten a la ecografía renal en el método imagenológico de primera elección para evaluar las anormalidades congénitas o adquiridas de los riñones (20).
El índice de Hodson es una ecuación matemática que permite estimar la longitud renal ideal para la talla de un niño. Se determina al aplicar la siguiente fórmula, aceptando una variación de +/- 2 como normal:
Índice de Hodson = (Talla cm X 0,057) + 2,646
Así, en el caso 1, con una talla de 96 cm, se espera una longitud renal de 8,1 cm. Sin embargo, la longitud renal de este paciente es de 5,1 cm para el riñón izquierdo y de 4,6 cm para el derecho. Así mismo, en el caso 2 el índice de Hodson es de 9,3 cm, mientras la longitud renal es 4,3 cm para el riñón izquierdo y 6,7 cm para el derecho.
Durante el desarrollo embrionario normal, del conducto mesonéfrico (De Wolf) surge una evaginación que se introduce en el metanefros, denominada brote ureteral; esta, eventualmente, originará el uréter, la pelvis renal, los cálices mayores y menores y hasta tres millones te túbulos colectores (21,22). El metanefros, por otro lado, originará los túbulos proximales y distales, así como los glomérulos (22). Las interacciones recíprocas a nivel celular entre el brote ureteral y el mesodermo metanéfrico que lo rodea condicionan el correcto desarrollo de las vías urinarias y de los riñones (23,24).
Las anomalías en las interacciones entre el brote ureteral y el metanefros (muchas de ellas posteriores al brote ectópico del brote ureteral), llevan a la mayoría de las ACRVU, incluyendo la hipoplasia renal (25). Algunos de los genes más importantes involucrados en este proceso de interacción en la nefrogénesis son aquellos que codifican para el factor neurotrófico derivado de la glía, expresado en el mesénquima del metanefros, y para su receptor a1; su mutación es una importante productora de HRC (10,26).
Otros genes expresados en el riñón primitivo y cuyas mutaciones pueden llevar a HRC, probablemente por alteración de las interacciones celulares entre el brote ureteral y el mesodermo metanéfrico que lo rodea, son los siguientes: gen PAX2, cuya mutación puede producir hipoplasia renal aislada o como parte del síndrome renal-coloboma (27-31); gen HNF1B (29,32); genes EYA1 (33) y SIX1 (30), que producen hipoplasia renal como parte del síndrome branquio-oto-renal (34); gen SALL1 (30), su mutación produce el síndrome de Townes-Brocks y entre las estructuras que afecta puede incluir al riñón en forma de hipoplasia renal (35); gen de la proteína morfogenética ósea 4 (25,36); gen del receptor tipo 2 de la angiotensina II (AGTR2) (25,37,38).
Adicionalmente, en el estudio de Sanna-Cherchi y cols., se encontraron más de 70 desórdenes genéticos diferentes asociados a malformaciones renales que incluían HRC (32), lo que demuestra la gran cantidad de alteraciones genéticas que pueden afectar la correcta nefrogénesis. Sin embargo, es muy variable la posibilidad de estos genes de desarrollar mutaciones; por ejemplo, es muy alta la frecuencia de mutaciones de EYA1 o PAX2 y muy baja en las mutaciones de AGTR2 (25). De lo anterior se deduce que la hipoplasia renal congénita puede atribuirse en su mayoría a la acumulación de pequeñas mutaciones en distintos genes, cada uno de los cuales tiene una función ontogénica en el riñón. Por otro lado, ciertas poblaciones, como la japonesa (39), exhiben una incidencia particularmente alta de hipoplasia renal congénita.
Toda esta evidencia, a la luz del caso reportado, sugiere que la afección en hermanos no es fruto de la coincidencia, sino que este trastorno tiene un importante trasfondo genético. Incluso, puede suceder que la HRC reportada en estos hermanos pueda afectar la descendencia de alguno de ellos o haya afectado ya a algunos de sus familiares, como sucedió en la familia reportada por Kerecuk y cols., en la que muchos parientes tenían hipoplasia o displasia renal congénita (40).
Por otro lado, en el estudio de Peters y cois., se evidenció que la obstrucción ureteral unilateral en el feto en desarrollo siempre se asociaba a disminución en el tamaño del riñón ipsilateral (41). No obstante, el riñón contralateral a la obstrucción era de tamaño considerablemente mayor como mecanismo compensatorio, mientras que, en el caso presentado, ambos riñones eran hipoplásicos. Por otro lado, en el estudio de Peters y cois., la obstrucción uretral se asociaba con hidronefrosis bilateral y no con hipoplasia renal bilateral (41). Adicionalmente, no hay evidencia de obstrucción de las vías urinarias en ninguno de los hermanos del caso reportado. No es factible entonces atribuir la hipoplasia renal bilateral de este caso a una obstrucción en el tracto urinario.
Las diferencias en el nivel de compromiso de la función renal y en el pronóstico de ambos niños fueron bastante llamativas, dado que el caso dos se encuentra con una función renal de más del doble del caso uno, a pesar de tener también HRC bilateral. Esto es lo usual en los casos familiares de HRC, como se evidenció por la gran variabilidad de presentaciones clínicas y de severidad de HRC entre familiares reportados por Kerecuk y cois., (40).
Para comprender estas variaciones se deben tener presente tres conceptos. Primero, el hecho de que el tamaño renal por ecografía no habla necesariamente del nivel de compromiso funcional renal ni de la cantidad de nefronas funcionales en pacientes con HRC, de modo que un riñón de menor tamaño puede perfectamente tener una mayor cantidad de estas nefronas que uno de mayor tamaño. Segundo, la figura de los "genes modificadores": se trata de genes que cambian la expresión de algunos otros. Por ejemplo, en una familia se evidenció que la mutación de PAX2 se asociaba a enfermedad renal cuando existían variaciones en la secuencia de SIX1 (30). En otros estudios se demostró que la mutación concomitante de dos genes diferentes se asocia con el desarrollo de enfermedad renal congénita (42,43). De este modo, diferencias en las mutaciones de múltiples genes entre los hermanos que componen los casos reportados pueden causar diferentes niveles de compromiso de la función renal y ser las causantes de las diferencias en las manifestaciones clínicas de su enfermedad.
Finalmente, se debe tener en mente el hecho de que las variaciones ambientales pueden influir en el desarrollo de los riñones de un feto en formación. Esto se ha evidenciado en modelos animales en los que la cantidad de proteína ingerida por la madre gestante modifica la expresión de genes y el desarrollo del metanefros (44). Así, aspectos desconocidos en el ambiente y durante la gestación de cada uno de los casos reportados pudieron también haber influido en la embriogénesis de sus tractos urinarios y, de esta manera, haber contribuido a la diferencia evidenciada en sus cuadros clínicos.
En el estudio de Sanna-Cherchi y cols., se encontró que la HRC bilateral era la anomalía renal congénita que más se diagnosticaba antes o al momento del nacimiento: más del 70% de los casos se diagnosticaron en estos dos períodos (6); es un hallazgo que no se dio en ninguno de los dos casos de este artículo, posiblemente por la no expresión clínica de esta patología.
Lo que eventualmente encaminó al diagnóstico de compromiso renal en el caso uno fue el estudio de síndrome de talla baja. Por lo tanto, es importante tener presente esta anomalía asociada a ERC como una de las posibles causas de un niño con síndrome de talla baja y, aun más importante, cuando tiene antecedente familiar de HRC. Esto se ve en el caso 2, quien, aun cuando se evidenciaba una talla considerablemente inferior al promedio y un hermano diagnosticado con HRC, se retrasó su diagnóstico dos años hasta que eventualmente presentó evidencia paraclínica de compromiso de la función renal.
Por lo inusual de la HRC y la poca evidencia de orientación para su diagnóstico, cobra importancia la información obtenida de estos dos casos, información que se comparte con la consciencia de que se requiere un estudio con otro diseño metodológico y multicéntrico: 1. Dado que la HRC es más prevalente en hombres que en mujeres (1,6), es coherente proponer que se tenga presente esta patología en hermanos sobre todo de sexo masculino de niños con HRC. 2. Sospechar la presencia de HRC en el hermano de un paciente afectado por esta anomalía, aun más cuando tiene manifestaciones de ERC como baja talla o una función renal disminuida, como se evidenció en el caso dos. 3. Quizás también el hecho de que el compromiso renal sea bilateral podría orientar en la sospecha; sin embargo, esto no puede deducirse a partir de un solo caso como el reportado. 4. Adicionalmente, si bien la madre poseía unos riñones de tamaño normal, este dato por sí mismo no conduce a descartar la posibilidad de HRC en la madre u otros familiares de futuros niños con esta afección.
Por otro lado en el estudio de Sanna-Cherchi y cois., se encontró que la probabilidad de supervivencia renal en casos de HRC a 10, 20 y 30 años de edad era de 100%, 84% y 70%, respectivamente (6), lo que les confiere un pronóstico reservado a largo plazo a los niños del caso reportado. Esta situación se resalta, particularmente, para el caso 1, que maneja cifras considerablemente menores de TFG y que se encuentra ya en lista de espera para trasplante renal.
Frecuentemente se presenta más de una ACRVU en un mismo sujeto (25); Para el caso de la HRC se ha encontrado asociación a obstrucción de la unión pieloureteral o reflujo vescicoureteral contralateral o ipsilateralmente (25,45). Este no fue el caso de ninguno de los dos hermanos que reportamos. Sin embargo la HRC bilateral en ellos se asoció a compromiso en la función renal.
Sobre el tratamiento de estos niños vale la pena resaltar que la terapia con Inhibidor de la Enzima Convertidora de Angiotensina (IECA), la cual tiene como objetivo brindar nefroprotección, aun cuando no modifica la historia natural de la HRC (46). El uso de esta terapia con miras a brindar nefroprotección es una práctica usual en estos casos, como se evidenció en el estudio observacional de Sanna-Cherchi y cois.; en dicho estudio, el 21% de los pacientes con HRC bilateral inició terapia con IECA al año del diagnóstico (6).
Conclusiones
El antecedente familiar de HRC en un paciente con compromiso de la función renal (que, como lo recuerdan estos casos puede manifestarse inicialmente con talla baja), debe conducir a sospechar la presencia de esta entidad y evaluarla mediante una ecografía renal y de vías urinarias. Por otro lado, si bien futuros estudios deberán establecer el porcentaje de evolución de HRC a ERC y el riesgo de HRC entre hermanos y familiares de pacientes afectados, principalmente cuando la afección es bilateral, pueden hacerse algunas recomendaciones en cuanto al manejo de los pacientes con HRC.
Los niños afectados, en especial aquellos con compromiso bilateral, pueden evolucionar a un compromiso de la función renal a largo plazo y terminar en ERC, razón por la cual en estos pacientes debe implementarse un programa de control de la función renal. Debería considerarse también la realización de ecografía renal y de vías urinarias a padres y hermanos de pacientes con esta entidad para detectar algún grado de compromiso familiar e iniciar controles y manejo precozmente. Finalmente, ante la complejidad de esta entidad es necesario que en su manejo participe un grupo multidisciplinario de profesionales que incluya médicos generales, pediatras, nefrólogos pediatras, radiólogos y genetistas.
Conflicto de intereses: Ninguno declarado por los autores.
Financiación: Ninguna declarado por los autores.
Agradecimientos: Ninguno declarado por los autores.
Referencias
1. Scott JE, Renwick M Urological anomalies in the Northern Region Fetal Abnormality Survey. Arch Dis Child. 1993;68:22-6. [ Links ]
2. Pope JC, Brock JW, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol. 1999;10:2018-28. [ Links ]
3. Birth Defects Monitoring Program (BDMP)/Commission on Professional and Hospital Activities (CPHA) surveillance data, 1988-1991. Teratology. 1993;48:658-75. [ Links ]
4. Livera LN, Brookfield DS, Egginton JA, Hawnaur JM. Antenatal ultrasonography to detect fetal renal abnormalities: a prospective screening programme. BMJ. 1989;298:1421-3. [ Links ]
5. Schulman J, Edmonds LD, McClearn AB, Jensvold N, Shaw GM Surveillance for and comparison of birth defect prevalences in two geographic areas--United States, 1983-88. MMWR CDC Surveill Summ. 1993;42:1-7. [ Links ]
6. Sanna-Cherchi S, Ravani P, Corbani V, Parodi S, Haupt R, Piaggio G et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76:528-33. [ Links ]
7. Loane M, Dolk H, Kelly A, Teljeur C, Greenlees R, Densem J,et al. Paper 4: EUROCAT statistical monitoring: identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res A Clin Mol Teratol. 2011;91:S31-43. [ Links ]
8. Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. 2012;27:363-73. [ Links ]
9. Warady BA, Hébert D, Sullivan EK, Alexander SR, Tejani A. Renal transplantation, chronic dialysis, and chronic renal insufficiency in children and adolescents. The 1995 Annual Report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol. 1997;11:49-64. [ Links ]
10. Mantan M, Sethi GR. Congenital anomalies of kidney and urinary tract in siblings: An uncommon condition. Indian J Nephrol. 2013;23:217-9. [ Links ]
11. Manouvrier-Hanu S, Devisme L, Farre I, Hue V, Storme L, Kacet N et al. Pulmonary hypertension of the newborn and urogenital anomalies in two male siblings: a new family with misalignment of pulmonary vessels. Genet Couns. 1996;7:249-55. [ Links ]
12. Hanlon-Lundberg KM, Verp MS, Loy G. Posterior urethral valves in successive generations. Am J Perinatol. 1994;11:37-9. [ Links ]
13. Schreuder MF, van der Horst HJ, Bokenkamp A, Beckers GM, van Wijk JA. Posterior urethral valves in three siblings: a case report and review of the literature. Birth Defects Res A Clin Mol Teratol. 2008;82:232-5. [ Links ]
14. Wiesel A, Queisser-Luft A, Clementi M, Bianca S, Stoll C, Group ES. Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: an analysis of 709,030 births in 12 European countries. Eur J Med Genet. 2005;48:131-44. [ Links ]
15. Hiraoka M, Hori C, Tsukahara H, Kasuga K, Ishihara Y, Sudo M. Congenitally small kidneys with reflux as a common cause of nephropathy in boys. Kidney Int. 1997;52:811-6. [ Links ]
16. Woolf AS. Renal hypoplasia and dysplasia: starting to put the puzzle together. J Am Soc Nephrol. 2006;17:2647-9. [ Links ]
17. Rosenbaum DM, Korngold E, Teele RL. Sonographic assessment of renal length in normal children. AJR Am J Roentgenol. 1984;142:467-9. [ Links ]
18. Emamian SA, Nielsen MB, Pedersen JF, Ytte L. Kidney dimensions at sonography: correlation with age, sex, and habitus in 665 adult volunteers. AJR Am J Roentgenol. 1993;160:83-6. [ Links ]
19. Jones TB, Riddick LR, Harpen MD, Dubuisson RL, Samuels D. Ultrasonographic determination of renal mass and renal volume. J Ultrasound Med. 1983;2:151-4. [ Links ]
20. Khazaei MR, Mackie F, Rosenberg AR, Kainer G. Renal length discrepancy by ultrasound is a reliable predictor of an abnormal DMSA scan in children. Pediatr Nephrol. 2008;23:99-105. [ Links ]
21. Sadler TW. Langman Embriología Médica. Décima edición. Buenos Aires: Editorial Médica Panamericana; 2008. [ Links ]
22. Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nat Rev Genet. 2002;3:533-43. [ Links ]
23. Saxén L, Sariola H. Early organogenesis of the kidney. Pediatr Nephrol. 1987;1:385-92. [ Links ]
24. Barasch J, Yang J, Ware CB, Taga T, Yoshida K, Erdjument-Bromage H et al. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell. 1999;99:377-86. [ Links ]
25. Ichikawa I, Kuwayama F, Pope JC, Stephens FD, Miyazaki Y. Paradigm shift from classic anatomic theories to contemporary cell biological views of CAKUT. Kidney Int. 2002;61:889-98. [ Links ]
26. Chen F. Genetic and developmental basis for urinary tract obstruction. Pediatr Nephrol. 2009;24:1621-32. [ Links ]
27. Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358-64. [ Links ]
28. Eccles MR, Schimmenti LA. Renal-coloboma syndrome: a multi-system developmental disorder caused by PAX2 mutations. Clin Genet. 1999;56:1-9. [ Links ]
29. Thomas R, Sanna-Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol. 2011;26:897-903. [ Links ]
30. Weber S, Moriniere V, Knüppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17:2864-70. [ Links ]
31. Torban E, Goodyer PR. Effects of PAX2 expression in a human fetal kidney (HEK293) cell line. Biochim Biophys Acta. 1998;1401:53-62. [ Links ]
32. Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet. 2012;91:987-97. [ Links ]
33. Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997;15:157-64. [ Links ]
34. Chen A, Francis M, Ni L, Cremers CW, Kimberling WJ, Sato Y, et al. Phenotypic manifestations of branchio-oto-renal syndrome. Am J Med Genet. 1995;58:365-70. [ Links ]
35. O'Callaghan M, Young ID. The Townes-Brocks syndrome. J Med Genet. 1990;27:457-61. [ Links ]
36. Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest. 2000;105:863-73. [ Links ]
37. Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2011;26:353-64. [ Links ]
38. Nishimura H, Yerkes E, Hohenfellner K, Miyazaki Y, Ma J, Hunley TE, et al. Role of the angiotensin type 2 receptor gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell. 1999;3:1-10. [ Links ]
39. Hiraoka M, Taniguchi T, Nakai H, Kino M, Okada Y, Tanizawa A, et al. No evidence for AT2R gene derangement in human urinary tract anomalies. Kidney Int. 2001;59:1244-9. [ Links ]
40. Kerecuk L, Sajoo A, McGregor L, Berg J, Haq MR, Sebire NJ, et al. Autosomal dominant inheritance of non-syndromic renal hypoplasia and dysplasia: dramatic variation in clinical severity in a single kindred. Nephrol Dial Transplant. 2007;22:259-63. [ Links ]
41. Peters CA, Carr MC, Lais A, Retik AB, Mandell J. The response of the fetal kidney to obstruction. J Urol. 1992;148:503-9. [ Links ]
42. Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet. 2002;11:379-88. [ Links ]
43. Badano JL, Kim JC, Hoskins BE, Lewis RA, Ansley SJ, Cutler DJ, et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12:1651-9. [ Links ]
44. Welham SJ, Riley PR, Wade A, Hubank M, Woolf AS. Maternal diet programs embryonic kidney gene expression. Physiol Genomics. 2005;22:48-56. [ Links ]
45. Atiyeh B, Husmann D, Baum M. Contralateral renal abnormalities in multicystic-dysplastic kidney disease. J Pediatr. 1992;121:65-7. [ Links ]
46. Ardissino G, Viganô S, Testa S, Daccô V, Paglialonga F, Leoni A, et al. No clear evidence of ACEi efficacy on the progression of chronic kidney disease in children with hypodysplastic nephropathy--report from the ItalKid Project database. Nephrol Dial Transplant. 2007;22:2525-30. [ Links ]
