










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.62 no.2 Bogotá Apr./June 2014
https://doi.org/10.15446/revfacmed.v62n2.45420
http://dx.doi.org/10.15446/revfacmed.v62n2.45420
Casos clínicos
Galactosemia como causa de ictericia neonatal
Neonatal jaundice caused by galactosemia
Mery Yolanda Cifuentes-Cifuentes1,2; Antonio Estrada-Vizcaíno2; Rosa Romero C3
1 División de Neonatología. Departamento de Pediatría. Facultad de Medicina. Universidad Nacional de Colombia. Bogotá, Colombia.
2 Instituto Materno Infantil. Hospital La Victoria. Bogotá, Colombia.
3 Servicio de Genética. Hospital La Victoria. Bogotá, Colombia.
Correspondencia: Mery Yolanda Cifuentes-Cifuentes. Dirección: División de Neonatología. Departamento de Pediatría. Facultad de Medicina, Universidad Nacional de Colombia. Carrera 10 No. 1-66 sur. Teléfono:+57 12338979.
Correo electrónico: mycifuentesd@unal.edu.co
Cifuentes-Cifuentes MY, Estrada-Vizcaíno A, Romero R. Galactosemia como causa de ictericia neonatal. rev.fac.med. 2014;62: 287-292.
Recibido: 18/04/2013 / Aceptado: 27/05/2014
Resumen
La ictericia es un problema muy frecuente en las unidades de recién nacidos. En este documento se presenta el caso de un neonato que cursó con hiperbilirrubinemia, inicialmente con predominio de la bilirrubina indirecta y posteriormente con anemia, hepatomegalia, aumento de la bilirrubina directa, alteración de la función hepática, disfunción tubular renal, cataratas y lesiones en la sustancia blanca del SNC.
Se descartaron diversas causas de hiperbilirrubinemia directa en el neonato. Se encontró positiva una prueba de azúcares reductores en orina que resultó ser galactosa en la cromatografía en capa fina. Se documentó marcada reducción de la actividad enzimática de galactosa uridil transferasa, con lo cual se confirmó el diagnóstico de galactosemia. Se suspendió la alimentación con leche materna, se inició leche de soya con resolución del cuadro clínico.
Palabras clave: Ictericia, Recién Nacido, Catarata, Galactosemias (DeCS).
Summary
Jaundice is a commonly occurring problem in hospitals' neonatal units. This report describes a newborn with hypefbilirrubinaemia; the clinical picture was initially dominated by the unconjugated bilirubin fraction and later by anaemia, hepatomegaly, increased conjugated bilirubin fraction, impaired liver function, renal tubular dysfunction, cataracts and injury to CNS white matter. Various causes of direct hyperbilirrubinaemia in the newborn were ruled out. Thin-layer chromatography revealed urine-reducing substances (i.e. galactose). A marked reduction in galactose uridyl transferaseactivity was documented, thereby confirming a diagnosis of galactosaemia. Breastfeedingwas discontinued and soy milk started which led to the symptoms becoming resolved.
Key words: Jaundice; Infant, Newborn; Cataract; Galactosemias (MeSH).
Introducción
Cerca del 70% de los recién nacidos presentan ictericia, frecuentemente por aumento de bilirrubina indirecta (no conjugada), lo cual origina un desequilibrio entre la producción de bilirrubina y su metabolismo; esta situación, de curso benigno, ha sido denominada ictericia fisiológica (1). La ictericia se considera patológica si se presenta en las primeras 24 horas de vida, si la cifra de bilirrubina aumenta más de 5 mg/ dL por día, si supera los 17 mg/dL o la fracción de bilirrubina directa (conjugada) es mayor al 20% del total de bilirrubina (2).
Múltiples patologías originan ictericia neonatal, unas cursan con aumento de bilirrubina indirecta: infección, sangrado oculto, policitemia, aumento de la circulación enterohepática, deficiencias en la conjugación (Crigler Najjar, Gilbert, hipotiroidismo) y procesos hemolíticos de diversa etiología (medicamentos como la estreptomicina y la vitamina K, isoinmunización Rh y ABO, defectos de morfología globular como la esferocitosis o la eliptocitosis, defectos enzimáticos como la deficiencia de piruvato kinasa, deficiencia de glucosa 6P-dehidrogenasa, deficiencia de galactosa-uridiltransferasa, deficiencia de glutatión, y defectos en la hemoglobina) (2-6).
Otras cursan con aumento de bilirrubina directa: obstrucción de la vía biliar, síndrome de Dubin-Johnson, síndrome de Rotor, infección urinaria, sepsis, infección intrauterina (TORCH), enfermedades metabólicas (deficiencia de alfa-antitripsina, fibrosis quística, galactosemia, glicogenosis, enfermedad de Wilson, enfermedad de Niemann-Pick, enfermedad de Gaucher), cromosomopatías y administración de medicamentos (acetaminofen, sulfa, rifampicina, tetraciclina, corticoides, eritromicina) (2).
La galactosemia, causa de hiperbilirrubinemia indirecta y también de hiperbilirrubinemia directa, es una enfermedad que se transmite con carácter autosómico recesivo, debida a la deficiencia de cualquiera de las tres enzimas de la vía de Leloir, encargada de convertir la galactosa en glucosa. La a-D-galactosa es fosforilada mediante la galactokinasa (GALK) a galactosa-1-fosfato; la galactosa-1-fosfato uridil transferasa (GALT) (a través del sustrato de la UDP glucosa) transforma la galactosa-1-fosfato en glucosa-1-fosfato y UDP galactosa, esta última, mediante la UDP-galactosa 4-epimerasa (GALE), es transformada en UDP glucosa (7).
La galactosemia clásica es causada por la deficiencia de galactosa-1-fosfato uridiltransferasa; la frecuencia varía entre 1:59.000 nacimientos en EEUU (8), 1:400.000 en Taiwán (9) y 1:9.000 en un estudio en Grecia (10). Actualmente, la galactosemia es objeto de tamizaje neonatal a nivel mundial (11). En Canadá, donde el tamizaje se hace desde 1984, la incidencia está entre 1:35.000 a 1:73.296 nacimientos (12).
El gen GALT ha sido mapeado en la región 9p13. La mutación Q188R cuenta para el 70% de las mutaciones deletéreas y conduce a una completa ausencia de actividad enzimática y al cuadro clásico de galactosemia (13).
Las manifestaciones clínicas incluyen: intolerancia a la alimentación, falla en el crecimiento, ictericia, daño hepático hasta cirrosis, tubulopatía renal, retardo mental y cataratas (14,15).
La sepsis por E. coli es frecuente (16-18), habiéndose demostrado disminución en la quimiotaxis y en la actividad bactericida de los neutrófilos (14,19). En pacientes con galactosemia se ha informado sepsis por Cándida, corea, púrpura fulminans, síndrome hemofagocítico y hemorragia vitrea (20-24).
En el laboratorio se puede encontrar: proteinuria, hiperbilirrubinemia con aumento de la indirecta inicialmente y luego de la directa, disminución de hemoglobina y hematocrito, aumento de aminotransferasas, prolongación de tiempos de coagulación, disminución de fibrinógeno, hipoglicemia, acidosis metabólica hiperclorémica, hipofosfatemia, sustancias reductoras en orina, aminociduria generalizada (25).
En la RMN de cerebro se encuentran lesiones en la sustancia blanca y en la espectroscopia se puede demostrar la acumulación de galactitol (26, 27). La entidad se sospecha por la presencia de sustancias reductoras en orina, lo que puede evidenciar la presencia de galactosa en la cromatografía de carbohidratos. Los niveles de galactosa en plasma y de galactosa-1-fosfato en eritrocitos se encuentran aumentados, los niveles de galactitol están aumentados en plasma y orina (7). La deficiencia de GALT se puede demostrar en los eritrocitos.
Es necesario realizar diagnóstico enzimático para descartar otras causas que originan galactosuria en el neonato: galactosemia transitoria por inmadurez hepática, hepatitis congénita, shunt portovenoso, tirosinemia tipo I, citrulinemia II (25). El diagnóstico prenatal se puede hacer por determinación enzimática en vellosidad corial o en cultivo de amniocitos o por cuantificación de galactitol en líquido amniótico (28). Existe evidencia de que en la deficiencia de GALT hay acumulación de galactosa, galactosa-1-fosfato y galactitol en el feto a partir de la semana 20 de gestación (29).
El tratamiento consiste en eliminar la galactosa de la dieta, cuya principal fuente es la leche y sus derivados; en el neonato con síntomas, debe suspenderse la leche materna y suministrarse leche de soya incluso antes de confirmar el diagnóstico (7).
El pronóstico a largo plazo es bueno para la supervivencia; sin embargo, se ha encontrado que, a pesar de haber suministrado dieta sin lactosa desde el nacimiento, se presenta daño a nivel del lenguaje, alteración motora y disfunción ovárica (30,31).
Presentación del caso
Recién nacido a término, masculino, peso 3.380 g (>°50), talla 51 cm (>°75), fruto de la primera gestación, padres no consanguíneos, madre adolescente, con control prenatal, O+, serología para VIH negativa, VDRL no reactiva; nacimiento vaginal institucional. Fue llevado a consulta a los 4 días de vida por presentar ictericia desde el primer día de vida; lo hospitalizaron e iniciaron fototerapia. Los laboratorios mostraron: hemoglobina 16 g, hematocrito 50%, leucocitos 15.700xmm3, PMN 65%, linfocitos 23%, plaquetas 167.000xmm3, O+, coombs negativo, reticulocitos 4,1%, bilirrubina directa 2,33 mg/dl, bilirrubina indirecta 14,18 mg/dl.
En los días siguientes hubo aumento de la cifra de bilirrubina total con aumento de la fracción conjugada, glicemia 55 mg%, aspartato aminotransferasa (AST) 86 UI/L, alanino aminotransferasa (ALT) 220 UI/L, fosfatasa alcalina 2741 U/L, tiempo de protrombina (TP) 1 min 5 seg, tiempo parcial de tromboplastina (TPT) mayor a 120 seg. Parcial de orina: densidad 1015, pH 7, proteínas >300 mg, cetonas indicios, Hb +++, pigmentos biliares +++, hematíes incontables. Con estos hallazgos remitieron al Instituto Materno Infantil a los ocho días de vida.
Al ingreso, la madre refirió anorexia, somnolencia y succión pobre; durante el examen se encontró: peso 3.100 g, ictérico, hipotónico, con hepatomegalia, DHT GI. Se iniciaron líquidos, se tomaron hemocultivos y, por sospecha de infección, se iniciaron ampicilina más gentamicina.
El cuadro hemático mostró hemoglobina 14 g, hematocrito 39%, leucocitos 10.570xmm3, PMN 61,6%, linfocitos 27%, monocitos 11%, plaquetas 244.000xmm3, anisocitosis en el extendido de sangre periférica, glicemia 54 mg%, gases sanguíneos: pH 7,32, pCO2 27, pO2 30, lactato 3,5, HCO3 13.9, BE -12, Na 138 mEq/L, Cl 111,2 mEq/L, Anion gap 12.9, AST 85 UI/L, ALT 165 UI/L, Bilirrubina total 27,7 mg/ dl, bilirrubina directa 5,43 mg/dl, fosfatasa alcalina 696 U/L, PCR<10, PT 39,3 seg, PTT 100,7 seg, parcial de orina: pH 7, proteínas ++, sangre +++, bilirrubinas ++. Un control al día siguiente mostró: bilirrubina total 18,03 mg/dl, bilirrubina directa 7,54 mg/dl (Figura 1).
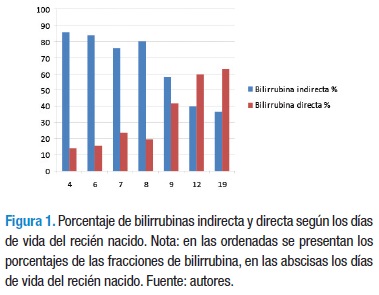
Los laboratorios mostraron TSH 0,075 microUI/ml, T4 libre 1,08 ng/dl, Ig M para toxoplasma, citomegalovirus, rubeola, herpes I y II negativas, antígeno de superficie para hepatitis B negativo, VDRL no reactiva, hemocultivos negativos. A los 12 días de vida los datos arrojaron: hemoglobina 10 g, hematocrito 28%, bilirrubina total 7,2 mg/dl, bilirrubina directa 4,3 mg/dl, PCR 31, amonio 0,44 micromol/L (VR 0,17-0,80).
Se realizó ecografía abdominal que no evidenció obstrucción de la vía biliar.
El estudio metabólico en orina informó: Benedict +, glucosa oxidasa -, Dinitrofenil hidracina (DNFH) +, Cloruro férrico (ClFe) -, Nitroprusiato (NP) -, Nitrosonaftol (NS) -. Banda sin identificar en cromatografía de aminoácidos en capa fina; en la cromatografía de carbohidratos en capa fina (Figura 2) se observó banda que migra a la altura de la galactosa.
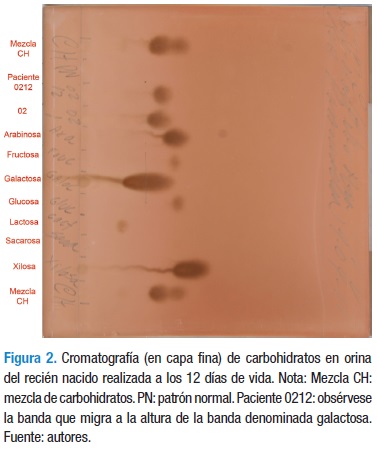
Se suspendió la leche materna y de fórmula y se suministró leche de soya. En los siguientes días se observó disminución de la ictericia y de la hepatomegalia, así como mejoría del tono muscular, con buen patrón de alimentación. Se tomó muestra de sangre en papel de filtro para perfil de acilcarnitinas, cuantificación de aminoácidos y de galactosa-1-fosfato por espectrometría de masas en tandem.
A los 19 días de vida los resultados eran: hemoglobina 9,34 g, hematocrito 24,92%, AST 96,5 UI/L, ALT 49 Ul/L, bilirrubina total 3,59 mg/dl, bilirrubina directa 2,27 mg/dl, fosfatasa alcalina 263 U/L, se transfundieron glóbulos rojos empaquetados. Oftalmología detectó catarata bilateral mayor en ojo derecho. Al mes de vida se realizó resonancia magnética nuclear de cerebro (Figura 3).
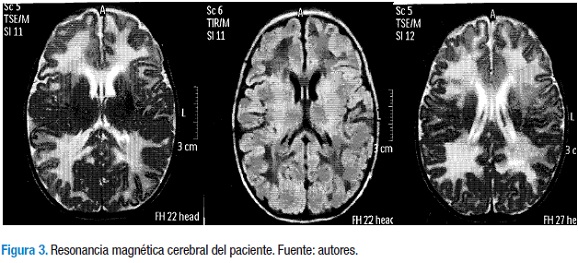
Se evidenció una marcada hipointensidad en T1 y FLAIR e hiperintensidad en T2 de la sustancia blanca peri y paraventricular, en relación con leucoencefalopatía probablemente de origen metabólico. Además, los datos arrojaron: AST 53,4 UI/L, ALT 30,9 UI/L, bilirrubina total 0,68 mg/dl, bilirrubina directa 0,34 mg/dl, PT 11,8 seg (control 13 seg), PTT 31,9 seg (control 27,8 seg). La muestra para cuantificación enzimática fue tomada después de haber administrado la transfusión sanguínea, por lo que se pospuso la determinación enzimática. De manera que se dio salida.
A los tres meses se realizó un control que arrojó los siguientes datos: peso 4.900 g (<°3), talla 57 cm (<°3), PC 41 cm (°50), hígado dos cm por debajo del borde costal, no sostiene la cabeza, sonrisa social, hipertonía MMII, manos empuñadas, aumento tono distal. Por su parte, el Informe de Espectrometría de Masas en Tandem (MSMS) evidenció: citrulina 46,21 pM (rango de referencia 6-33), galactosa-1-fosfato en sangre 0,45 mmol/l (rango de referencia <0,50) (la muestra se tomó cuando el paciente ya tenía restricción de lactosa). Habilitación pediátrica lo valoró y encontró déficit severo del desarrollo. Se envió a nutrición, neuropediatría, terapia ocupacional y oftalmología.
Un nuevo control, a los nueve meses, presentó los siguientes resultados: peso 8.580 g (<°50), talla 69 cm (<°15), PC 46 cm (<°85). Vacunación acorde a la edad. Recibe leche de soya y complementaria. Se sienta, sonríe, coge el biberón, tiene dos dientes; no se encontró hepatomegalia. Resolución de las cataratas por informe verbal de oftalmología.
Se tomó muestra para determinación de actividad enzimática de GALT y gotas de sangre en papel filtro para cuantificación de galactosa-1-fosfato y de aminoácidos por MSMS que informaron: actividad enzimática GALT 0,82 UI/hora/ml (valor de referencia 3,5-14 UI/hora/ml); galactosa-1-fosfato en sangre 0,10 mmol/l, citrulina 14,78 pM por MSMS. Con los datos se confirmó el diagnóstico de deficiencia de GALT.
Discusión
Tal como ha sido informado en la literatura (5,25,32) y como ocurrió en un caso detectado en el Instituto Materno Infantil y publicado previamente (4) este paciente presentó inicialmente un cuadro de hiperbilirrubinemia indirecta, debida a la hemólisis ocasionada por acumulación de galactosa-1-fosfato en los eritrocitos y por inhibición de la conjugación de la bilirrubina que ocasiona este metabolito (5,14,33).
El cambio de hiperbilirrubinemia con predominio de la fracción no conjugada a hiperbilirrubinemia, con aumento de la fracción conjugada con hepatomegalia y disfunción hepática, obligó a descartar las principales causas de hiperbilirrubinemia directa en los neonatos: atresia u obstrucción de la vía biliar, sepsis, infección urinaria, sífilis, infección por citomegalovirus, toxoplasma, herpes I y II, hepatitis B y rubeola. El paciente no recibió medicamentos como rifampicina o eritromicina, descritas como causas de colestasis (2).
Se inició investigación para errores innatos del metabolismo. La sospecha de galactosemia se tuvo al demostrar la presencia de azúcares reductores en orina (prueba de Benedict), acompañada de una prueba para glucosa negativa (glucosa oxidasa) para, posteriormente, en la cromatografía en capa fina de carbohidratos en orina, encontrar la presencia de galactosa (34,35).
La presencia de cataratas (32), la leve hipoglicemia y las alteraciones en el parcial de orina que mostraron una tubulopatía proximal con acidosis metabólica hiperclorémica con anion gap normal (25) hacen parte del cuadro clínico de galactosemia clásica; la RMN cerebral mostró afectación de la sustancia blanca, con hallazgos idénticos a los informados por Otaduy en un paciente con galactosemia: marcada hipointensidad en T1 y FLAIR (fluid-attenuated inversion recovery) e hiperintensidad en T2 (26).
Con la sospecha diagnóstica se suprimió la galactosa de la dieta, se suministró leche de soya, con lo cual se obtuvo una mejoría dramática del cuadro clínico, tal como se informa en la literatura. Las fórmulas sin lactosa con hidrolizado de proteína no se deben usar pues contienen galactosa por tener trazas de caseína y seroalbúmina (36). El diagnóstico se confirmó al demostrar la disminución de la actividad de galactosa uridiltransferasa en glóbulos rojos, patrón de oro para el diagnóstico de la galactosemia clásica (32,35).
Era urgente la cuantificación enzimática por haberse encontrado una elevación moderada de citrulina, lo cual hizo necesario descartar una citrulinemia tipo II o una aciduria argininosuccínica. El cuadro clínico no era compatible con citrulinemia tipo II, pues el paciente no tenía restricción de crecimiento y el aumento de citrulina no se acompañaba de hipermetioninemia ni hiperfenilalaninemia, hallazgos característicos en estos pacientes (37). La elevación de la citrulina tampoco correspondía a una aciduria argininosuccínica, defecto del ciclo de la urea en cuyo curso clínico no se encuentra colestasis y cuyo principal hallazgo es la hiperamonemia. El paciente del caso documentado tenía una cuantificación de amonio normal (38,39).
La presentación del caso pretende enfatizar la necesidad de incluir la galactosemia en el espectro etiológico de la ictericia neonatal, ya sea que curse con aumento de la bilirrubina indirecta o con aumento de la bilirrubina directa. Para ello, se ha ilustrado la secuencia para su diagnóstico, dado que la galactosemia es una entidad potencialmente fatal en la infancia; si no se instaura el tratamiento puede ocurrir muerte por la misma enfermedad o por sepsis.
Conflicto de interés: Ninguno declarado por los autores.
Financiación: Ninguna declarada por los autores.
Agradecimientos: Ninguno declarado por los autores.
Referencias
1. Kaplan M, Muraca M, Hammerman C, Rubaltelli F, Vilei MT, Vreman HJ, Stevenson DK Imbalance between production and conjugation of bilirubin: a fundamental concept in the mechanism of neonatal jaundice. Pediatrics. 2002;110:e47. [ Links ]
2. Porter ML, Dennis BL. Hyperbilirubinemia in the term newborn. Am Fam Physician. 2002;65:599-606. [ Links ]
3. Abdel Fattah M, Abdel Ghany E, Adel A, Mosallam D, Kamal S. Glucose-6-phosphate dehydrogenase and red cell pyruvate kinase deficiency in neonatal jaundice cases in egypt. J Pediatr Hematol Oncol. 2010;27:262-71. [ Links ]
4. Cifuentes Y, Arteaga C, Bermúdez M, Quintero L. Ictericia e hipoglicemia neonatal: un caso de galactosemia. MEDICAS UIS. 2002;16:30-2. [ Links ]
5. Woo HC, Phornphutkul C, Laptook AR. Early and severe indirect hyperbilirubinemia as a manifestation of galactosemia. J Perinatol. 2010;30:295-7. [ Links ]
6. Cifuentes Y, Bermúdez M. Aciduria piroglutámica en un neonato con anemia hemolítica. rev.fac.med. 2000;48:195-8. [ Links ]
7. Baldellou Vásquez A, García Jiménez MC. Errores congénitos del metabolismo de la galactosa. En Sanjurjo P, Baldellou A (Eds). Diagnóstico y tratamiento de las enfermedades metabó-licas hereditarias. Madrid: Editorial Ergon; 2010:399-409. [ Links ]
8. Kwon C, Farrell PM. The magnitude and challenge of falsepositive newborn screening test results. Arch Pediatr Adolesc Med. 2000;154:714-8. [ Links ]
9. Cheung KL, Tang NL, Hsiao KJ, Law LK, Wong W, Ng PC, et al. Classical galactosaemia in Chinese: A case report and review of disease incidence. J Paediatr Child Health. 1999;35:399-400. [ Links ]
10. Schulpis K, Papakonstantinou ED, Michelakakis H, Pods-karbi T, Patsouras A, Shin Y. Screening for galactosaemia in Greece. Paediatr Perinat Epidemiol. 1997;11:436-40. [ Links ]
11. Schweitzer-Krantz S. Early diagnosis of inherited metabolic disorders towards improving outcome: the controversial issue of galactosaemia. Eur J Pediatr. 2003;162:S50-3. [ Links ]
12. Shah V, Friedman S, Moore AM, Platt BA, Feigenbaum ASJ. Selective screening for neonatal galactosemia: an alternative approach. Acta Paediatr. 2001;90:948-9. [ Links ]
13. Schuster V, Podskarbi T, Ottensmeier H, Haubner M, Shin YS. Simultaneous occurrence of various mutations and polymorphisms in cis and in trans of the galactose-1-phosphate uridyl-transferase gene in a Turkish family with classical galactosemia. J Mol Med (Berl). 1998;76:715-9. [ Links ]
14. Holton JB, Walter JH, Tyfield LA. Galactosemia. En Scriver CH, Beaudet A, Valle D, Sly W (Eds). The metabolic and molecular bases of inherited disease. New York, London: Mac Graw Hill Inc; 2001:1553-87. [ Links ]
15. Scheibenreiter S, Knoll E, Widhalm K Long-term results in children with classical galactosemia. Wien Klin Wochenschr. 1992;104:514-7. [ Links ]
16. Kumar M, Yachha SK, Gupta RK Neonatal cholestasis syndrome due to galactosemia. Indian J Gastroenterol. 1996;15:26-7. [ Links ]
17. Janaud JC, Ropert JC, Choulot JJ, Becart E, Dehan M, Gabilan JC. Congenital galactosemia detected by severe Escherichia coli infections. Arch Fr Pediatr. 1979;36:508-11. [ Links ]
18. Rathi N, Rathi A. Galactosemia presenting as recurrent sepsis. J Trop Pediatr. 2011;57:487-9. [ Links ]
19. Kobayashi RH, Kettelhut BV, Kobayashi AL. Galactose inhibition of neonatal neutrophil function. Pediatr Infect Dis. 1983;2:442-5. [ Links ]
20. Verma S, Bharti B, Inusha P. Association of Fungal Sepsis and Galactosemia. Indian J Pediatr. 2010;77:695-6. [ Links ]
21. Shah PA, Kuchhai FA. Galactosemia with Chorea -An unusual presentation. Indian J Pediatr. 2009;76:97-8. [ Links ]
22. Zenciroglu A, Ipek MS, Aydin M, Kara A, Okumus N, Kilic M. Purpura fulminans in a newborn infant with galactosemia. Eur J Pediatr. 2010;169:903-6. [ Links ]
23. Marcoux MO, Laporte-Turpin E, Alberge C, Fournie-Gardini E, Castex MP, Rolland M et al. Congenital galacto-saemia: an unusual presentation. Arch Pediatr. 2005;12:160-2. [ Links ]
24. Levy HL, Brown AE, Williams SE, de Juan E Jr. Vitreous hemorrhage as an ophthalmic complication of galactosemia. J Pediatr. 1996;129:922-5. [ Links ]
25. Berry GT. Galactosemia: When is it a newborn screening emergency? Mol Genet Metab. 2012;106:7-11. [ Links ]
26. Otaduy MC, Leite CC, Lacerda MT, Costa MO, Arita F, Prado E, Rosemberg S. Proton MR spectroscopy and imaging of a galactosemic patient before and after dietary treatment. AJNR Am J Neuroradiol. 2006;27:204-7. [ Links ]
27. Berry GT, Hunter JV, Wang Z, Dreha S, Mazur A, Brooks DG, et al. In vivo evidence of brain galactitol accumulation in an infant with galactosemia and encephalopathy. J Pediatr. 2001;138:260-2. [ Links ]
28. Jakobs C, Kleijer WJ, Allen J, Holton JB. Prenatal diagnosis of galactosemia. Eur J Pediatr. 1995;154:S33-6. [ Links ]
29. Holton JB. Effects of galactosemia in utero. Eur J Pedia-tr. 1995;154:S77-81. [ Links ]
30. Jumbo-Lucioni P, Garber K, Kiel J, Baric I, Berry GT, Bosch A, et al. Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. J Inherit Metab Dis. 2012;35:1037-49. [ Links ]
31. Schadewaldt P, Hoffmann B, Hammen HW, Kamp G, Schweitzer-Krantz S, Wendel U. Longitudinal assessment of intellectual achievement in patients with classical galactosemia. Pediatrics. 2010;125:e374-81. [ Links ]
32. Bosch AM. Classical galactosaemia revisited. J Inherit Metab Dis. 2006;29:516-25. [ Links ]
33. Burton BK. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics. 1998;102:E69. [ Links ]
34. Gutiérrez García E, Barrios García B, Carrillo Estrada U, Lantigua Cruz A. Galactosemia: diagnóstico de 5 casos con deficiencia de transferasa. Rev Cubana Pediatr. 2001;73:75-80. [ Links ]
35. Henderson H, Leisegang F, Brown R, Eley B. The clinical and molecular spectrum of galactosemia in patients from the Cape Town region of South Africa. BMC Pediatr. 2002;2:7. [ Links ]
36. Cornejo V, Raiman E. Alteración del metabolismo de la galactosa. Rev Chil Nutr [internet]. 2004;31(Suppl. 1):170-6. [citado 2014 abril 11]. Disponible en: http://dx.doi.org/10.4067/S0717-75182004031100004. [ Links ]
37. Ohura K, Kobayashi Y, Tazawa D, Abukawa O, Sakamoto S, Tsuchiya T et al. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis. 2007;30:139-44. [ Links ]
38. Cifuentes Y, Arteaga C, Lonngi G, Quintero L, Bermúdez M. Hiperamonemia neonatal causada por defectos del ciclo de la urea. rev.fac.med. 2010;58:316-23. [ Links ]
39. Beck NM, Johnston JP, Lemke KS, Pogacar P, Phornphutkul C. Rhode Island metabolic newborn screening: the effect of early identification. A case report of argininosuccinic aciduria (ASA). Med Health R I. 2011;94:121-3. [ Links ]
