










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.62 no.4 Bogotá Oct./Dec. 2014
https://doi.org/10.15446/revfacmed.v62n4.44015
DOI: http://dx.doi.org/10.15446/revfacmed.v62n4.44015
REVIEW ARTICLE
Nav1.5 cardiac sodium channels, regulation and clinical implications
Canales de sodio Nav1.5 cardíacos, regulación e implicaciones clínicas
Henry Humberto León-Ariza1,2, Natalia Valenzuela-Faccini3, Ariana Carolina Rojas-Ortega3, Daniel Alfonso Botero-Rosas1,4
1 Área de Morfofisiología, Facultad de Medicina, Universidad de La Sabana. Chía, Colombia.
2 Facultad de Enfermería, Fundación Universitaria Sanitas. Bogotá, Colombia.
3 Facultad de Medicina, Universidad de La Sabana. Chía, Colombia.
4 Facultad de Medicina, Universidad Antonio Nariño. Bogotá, Colombia.
Correspondence: Henry Humberto León-Ariza. Universidad de La Sabana, Campus del Puente del Común, Km. 7, Autopista Norte de Bogotá, Edificio F, Segundo piso. Chía, Cundinamarca, Colombia. Fax: +57 1 8616666. E-mail: henrylear@clinicaunisabana.edu.co.
Received: 13/06/2014 Accepted: 28/07/2014
Summary
Voltage-gated sodium channels constitute a group of membrane proteins widely distributed thought the body. In the heart, there are at least six different isoforms, being the Nav1.5 the most abundant. The channel is composed of an α subunit that is formed by four domains of six segments each, and four much smaller β subunits that provide stability and integrate other channels into the α subunit. The function of the Nav1.5 channel is modulated by intracellular cytoskeleton proteins, extracellular proteins, calcium concentration, free radicals, and medications, among other things. The study of the channel and its alterations has grown thanks to its association with pathogenic conditions such as Long QT syndrome, Brugada syndrome, atrial fibrillation, arrhythmogenic ventricular dysplasia and complications during ischemic processes.
Keywords: NAV1.5 Voltage-Gated Sodium Channel; Long QT syndrome; Brugada syndrome (MeSH).
León-Ariza HH, Valenzuela-Faccini N, Rojas-Ortega AC, Botero-Rosas DA. Nav1.5 cardiac sodium channels, regulation and clinical implications. Rev Fac Med. 2014;62(4):587-92. http://dx.doi.org/10.15446/revfacmed.v62n4.44015.
Resumen
Los canales de Sodio dependientes de voltaje constituyen un grupo de proteínas de membrana ampliamente distribuidas en el cuerpo humano. En el corazón se dispone de al menos seis diferentes isoformas de estos canales: los Nav1.5 son los más abundantes. El canal está constituido por una subunidad α, formada por cuatro dominios, cada uno de estos con seis segmentos y cuatro subunidades β mucho más pequeñas que estabilizan la estructura e integran la subunidad α de otros canales. La función del canal Nav1.5 se ve modulada por proteínas del citoesqueleto, proteínas extracelulares, concentraciones de calcio, radicales libres, medicamentos, entre otros. El estudio del canal y sus alteraciones se ha incrementado gracias a la asociación de este con condiciones patológicas como el síndrome de QT largo, el síndrome de Brugada, la fibrilación auricular, la displasia ventricular arritmogénica y por las complicaciones de en procesos isquémicos.
Palabras clave: Canal de Sodio Activado por Voltaje NAV1.5; Síndrome de QT Prolongado; Síndrome de Brugada (DeCS).
León-Ariza HH, Valenzuela-Faccini N, Rojas-Ortega AC, Botero-Rosas DA. Canales de sodio Nav1.5 cardíacos, regulación e i clínicas. Rev Fac Med. 2014;62(4):587-92. http://dx.doi.org/10.15446/revfacmed.v62n4.44015.
Introduction
The great majority of the cells in the organism need to be depolarized in order to fulfill their functions. This is the case for excitable tissues like neurons, and skeletal and cardiac muscle. Here, the sodium ion (Na+) is principally responsible for the process, as described by Hodgkin and Huxley in 1952 (1). Voltage-gated sodium channels, which differ in their alpha subunits (2), are responsible for Na+ flow. The difference in subunits of these channels has permitted their organization into isoforms of these channels that are mainly found in the central nervous system (Nav1.1 - 1.3 and Nav1.6), in the peripheral nervous system (Nav1.7 - 1.9), in skeletal muscle (Nav1.4), in myocardial muscle (Nav1.5) (3,4), and in the uterus, astrocytes and hypothalamus (Nax) (5). A large part of the classification process was possible thanks to the sensitivity of the proteins to tetradotoxin (TTX) (6,7), to which some Nav, like 1.5, 1.8 and 1.9, are more resistant and others, like Nav1.1 – 1.4 and 1.6, are more sensitive (3,8).
The cardiac muscle cells depend on a rapid influx of sodium (INa) to generate an action potential. This permits a depolarization of the tissue and conduction of the impulse through the myocardial cells (9,10). Until some time ago, it was thought that the INa in the heart depended solely on one type of channel (Nav1.5) (Figure 1). However, it is now clear that there are at least six different types of voltage-dependent sodium channels in cardiac contractile cells, Nav1.1 - 1.6, distributed at both the ventricular (11) and atrial levels (12). Furthermore, Nav1.8 channels have been also described that are related to neuronal regulation of the firing rate. They are located essentially in the electrical conduction system of the heart (13). Through the use of immunohistochemistry, it has been established that the Nav1.5 are especially located on the cellular surface of contractile, improving conduction between them. In addition, they are proportionally more abundant (88%) in the cellular surface (14-17).
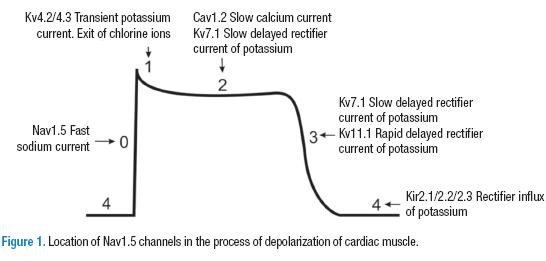
Due to the importance of the Nav1.5 channel, this review presents its fundamental characteristics and its principle physiological regulation mechanisms to finally link alterations of the channel with congenital and acquired cardiomyopathies.
The search for articles was carried out in the following databases: Pubmed, Sciencedirect and Scielo. The terms Nav1.5, Brugada syndrome, and atrial fibrillation (Nav1.5, síndrome de brugada, fibrilación auricular), in both English and Spanish, were used between January 2004 and February 2014 for the article search. Five articles from before 2004 were included because of their contribution to the theoretical construction.
Structure of the Nav1.5 channel
The Nav1.5 channel is a protein with a glycosylated membrane, codified by the gene SCN5A in the chromosome 3p21. It has a weight of 220 kDa and is made up of between 2014 y 2016 amino acid residues (14,18,19). This protein is formed by the homologous domains DI-DIV. Each one contains six transmembrane segments, S1-S6 (15). The α-subunit contains an amine group (N) and an intracellular carboxyl terminal domain (C), and three intracellular, called L1, L2, and L3 that connect the four domains (20). The most studied of the segments is S4, which is positively charged thanks to the presence of la arginine and lysine, followed by two hydrophobic residues. This segment is associated with the activation of the channel (21).
Furthermore, the inactivation of the channel is to be attributed to the DIII and DIV intracellular loops, where a sequence of three amino acids — isoleucine-phenylalanine-methionine— is fundamental. Also important in the inactivation is the carboxyl terminus sequence (14, 15). Between S5 and S6 of every domain is a loop that reintroduces itself in the lipid bilayer in the extracellular side of the pore formed by a narrow selective filter for the sodium ion, with two characteristic sequences of amino acids: aspartate-glutamate-lysine-alanine and glutamate-glutamate-aspartate-aspartate (6) (Figure 2).
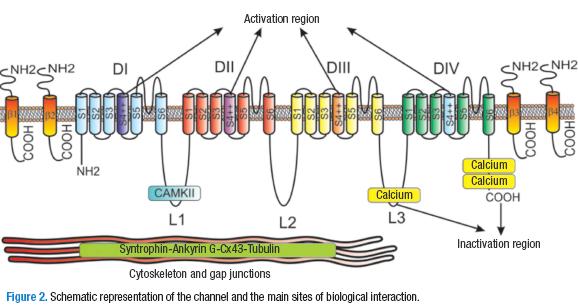
In addition to the α-subunit, the channel consists of the subunits β1-β4, which are proteins of 30-35 kDA (14). Its structure is characterized for being a transmembrane α-helix chain with a N-terminal extracellular fold similar to the one in immunoglobins and a short intracellular C terminus. The β1 and β3 subunits attach to the alpha-subunits in a non-covalent manner, while the β2 and β4 subunits attach through disulfide bonds. The function of these subunits is to modulate the opening and closing of the channel by interacting with the extracellular matrix and with the cytoskeleton at the same time. This influences the localization of these channels in the cell surface (12,16). Recently, it has been demonstrated that the β-1 subunit promotes interaction between the alpha subunits of the cardiac sodium channels Nav1.5 (20,22).
Biological characteristics of the Nav1.5 channel
As was previously mentions, the INa in the heart are produced mainly by the Nav1.5 channels, which are responsible for the creation of the action potential and the rapid depolarization of the heart (20,23). The Nav1.5 channels can be found in three states: closed, when the membrane has a resting potential; open, during depolarization; and inactive, during the state of non-conductivity. Inactivation is carried out thanks to an “inactivation gate” that is found in the intracellular loop that joins domains III and IV (6, 21). Furthermore, recovery in the inactivation state occurs during re-polarization in diastole (15).
Different experimental studies both in humans and animals have shown the presence of Nav1.5 channels not only at the level of cell membranes in cardiac muscle but also in intercalated discs, in lateral cell membranes, and in T-tubules (15). Also, the presence of these channels has been described in atrial myofibroblasts similar to those in excitable cells, which can modify their properties in pathological conditions (24). The Nav1.5 channels are distributed in an non-uniform manner along the ventricular wall, being more abundant in the endocardium than the epicardium, which generates important electrophysiological changes at the transmural level (25). This could explain the differences in the clinical behavior of subepicardial and subendocardial infarctions.
Interestingly, it has been found how sodium channels interact and are modulated by the mechanical properties of the membrane and cytoskeleton. This occurs because the Nav1.5 channels have a mechanosensitive property which means that the alterations in cellular mechanical behavior lead to changes at the channel level. For example the level of stretching produces directly proportional changes in its activation and inactivation (23). As such, changes at the mechanical level, like the behavior of blood volume (preload), lead to changes in the firing rate of the sinuatrial node, atrial-ventricular excitability, and conduction speed (26). Dystrophin and syntrophin, thanks to their relationship with the cytoskeleton, participate in the regulation of the Nav1.5, and are responsible for patients with muscular dystrophy presenting cardiopathies that are partially compensated for through utrophin (27,28).
Another widely recognized regulator of Nav1.5 channel function is calcium. This ion modulates the density of sodium channels without changes in the kinetic properties of the myocytes (29). The intracellular calcium level modulates that inactivation of the Nav1.5 in three regions of the channel: between the domains DIII and DIV, in two regions of the carboxyl terminus, and in isoleucine and glutamine sequences known as the IQ-motif (30). In addition, the calcium-calmodulin (CaM) interaction is fundamental for the activation of the calcium-calmodulin kinase (CAMKII), with which it interacts in the L1 region of the Nav1.5. Thus, the L1 domain is the predominant substrate for the CaMKII. 7 sites of phosphorylation have been found in 5 regions of the L1 domain (20,31). Although the results of the different studies are contradictory, it appears that the interaction of the Nav1.5 with the CAMKII slows the inactivation of the channel.
The intercalated disc serves as a meeting point of the proteins that are relevant for excitability, propagation, and the mechanical joining of cardiomyocytes. This converts it into a site of interaction between the sodium channels and the gap junctions. Specifically, we are referring to two interactions of proteins that relate the sodium channel and the gap junctions: conexin43 (Cx43) and ankyrin-G. This communication is important because it determines the intensity of the sodium current between the cardiomyocytes (32).
An increase in the polymerization of tubulin of the cytoskeleton, like the one caused by the anticarcinogenic agent taxol (TXL) reduces the expression of Nav1.5 in the sarcolemma, and reduces the INa in a process that may or may not involve the β1 subunit by producing a reduction in fast inactivation in the Nav1.5 + β1 interaction (33).
Another Nav1.5 modulator is the presence of nicotinamide adenine dinucleotide (NADH) through protein kinase C (PKC), in such a way that increases in NADH and free oxygen radicals (ROS) leads to the reduction of INa (34). Nav1.5 has also demonstrated its extrinsic regulation through medications: lidocaine is a blocker of Nav1.5, reason for which it is used as an antiarrhythmic (35); ranolazine blocks the mechanosensitivity of the channel and has and has an antianginal effect (26); propanolol —a beta-blocker— alters the cardiac sodium channel (36); and opioid medications interact with omega cardiac receptors that regulate the Nav1.5 while producing inhibition (35).
Many other structural and induced interactions between Nav1.5 and proteins have been described. For a more detailed review of these, we suggest consulting Rook et al (14).
Clinical application
The sodium channels Nav1.5 are, without a doubt, the most studied with regard to both congenital and acquired conditions. The first congenital pathological condition in which an association with mutations of the gene (SCN5A) was described was a form of the long QT syndrome (type 3) that predisposes sufferers to the development of ventricular tachycardia and torsades de pointes. In this case, the mutation of the channel leads to a slight prolongation of its action that delays the re-polarization phase (37). A similar phenomenon has also been described in the Brugada syndrome, which has a electrocardiographic pattern characteristic of supraunleveling for the ST segment in V1 to V3 and a morphology with right bundle branch block. It is related to sudden death (38). The prevalence of Brugada syndrome is not completely clear, since if may present in a silent or undiagnosed way. It has been estimated that there are around 5 cases per 10 000 people, and that it is the cause of death in 4-12% of sudden deaths (39).
Other pathological conditions associated with the Nav1.5 subunit include atrial fibrillation (AF). This is one of the most common persistent arrhythmias. It involves alterations to the sodium channel leading to an increase in conduction time that, in conditions of vagal stimulation, are prolonged even more, leading to the development of AF (40-44). It has also been reported that mutations of the β1 y β2 of the sodium channel are associated with AF and, in this case, a reduction in INa has been proposed as part of its pathogenesis (45). Mutations in the SCN5A gene have also been associated with the development of ventricular arrhythmogenic dysplasia and, thus, a greater tendency toward the development of ventricular arrhythmias like ventricular tachycardia and fibrillation (46-50).
The development of ventricular and atrial arrhythmias has been linked, in patients with heart failure, to the elevated presence of angiotensin II and hypoxia. These induce the activation of nuclear proteins like RBM25 and LUC7L3 that produce splicing in the SCN5A gene that is responsible for coding for the Nav1.5. This leads to a lesser expression of this protein and the development of arrhythmias (51).
In ischemic processes, extracellular acidosis interacts with Nav1.5, altering the states of rapid and slow inactivation. This increases the persistence of INa, something that contributes to the development of arrhythmias (52). Furthermore, recently the presence of coxsackievirus receptors can alter sodium currents in the Nav1.5, altering its function, which leads, once again, to a predisposition to the developments of arrhythmias in patients with coronary heart disease (53). Increases in NADH and ROS leads to a reduction in INa (blocks, arrhythmias), a commonly observed phenomenon in heart failure (34).
Conclusions
Nav1.5 sodium channels are no longer seen as a simple set of proteins that regulate sodium flow into the cell. Currently it is known that its function depends on the interaction with the cytoskeleton, second messengers, and other ions, such as calcium, that play an important role in pathological conditions. Much more research is needed in order to understand the biological nature of the Nav1.5 channel, which allows, among other applications, pharmacological development to prevent electrophysiological complications in acute myocardial infarction and to avoid the complications of conditions such as long QT syndrome or Brugada syndrome.
Conflict of interest
None declared by the authors.
Financing
None declared by the authors.
Acknowledgements
To the morphophysiology area of the Faculty of Medicine of the Universidad de La Sabana and the research incubator group for signal and images processing (PROSEIM).
References
1. Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. 2012; 590:2577-89. http://doi.org/zdx. [ Links ]
2. Savio-Galimberti E, Gollob MH, Darbar D. Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front Pharmacol. 2012; 3:124. http://doi.org/zdz. [ Links ]
3. Ogata N, Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn J Pharmacol. 2002;88(4):365-77. http://doi.org/bstv45. [ Links ]
4. Goldin AL. Evolution of voltage-gated Na(+) channels. J Exp Biol. 2002; 205:575-84. [ Links ]
5. García-Villegas R, López-Álvarez LE, Arni S, Rosenbaum T, Morales MA. Identification and functional characterization of the promoter of the mouse sodium-activated sodium channel Na(x) gene (Scn7a). J Neurosci Res. 2009; 87:2509-19. http://doi.org/fwv7zc. [ Links ]
6. Yu F, Catterall W. Overview of the voltage-gated sodium channel family. Genome Biol. 2003; 4:207.1-7. [ Links ]
7. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005; 57:411-25. http://doi.org/cxk7nr. [ Links ]
8. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000; 26:13-25. http://doi.org/dnt8s4. [ Links ]
9. Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013; 1833:886-94. http://doi.org/zd2. [ Links ]
10. Reinhard K, Rougier JS, Ogrodnik J, Abriel H. Electrophysiological properties of mouse and epitope-tagged human cardiac sodium channel Na v1.5 expressed in HEK293 cells. F1000Res. 2013;2:48. [ Links ]
11. Westenbroek RE, Bischoff S, Fu Y, Maier SK, Catterall WA, Scheuer T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J Mol Cell Cardiol. 2013; 64:69-78. http://doi.org/zd3. [ Links ]
12. Kaufmann SG, Westenbroek RE, Maass AH, Lange V, Renner A, Wischmeyer E, et al. Distribution and function of sodium channel subtypes in human atrial myocardium. J Mol Cell Cardiol. 2013; 61:133-41. http://doi.org/zd4. [ Links ]
13. Verkerk AO, Remme CA, Schumacher CA, Scicluna BP, Wolswinkel R, de Jonge B, Bezzina CR, et al. Functional Nav1.8 channels in intracardiac neurons: the link between SCN10A and cardiac electrophysiology. Circ Res. 2012; 111:333-43. http://doi.org/zd5. [ Links ]
14. Rook M, Evers M, Vos M, Bierhuizen M. Biology of Cardiac Sodium Channel Nav 1.5 Expression. Cardiovasc Res. 2012; 93:12-23. http://doi.org/bdsdkj. [ Links ]
15. Huges A. Roles and regulation of cardiac sodium channel Nav 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007; 76:381-9. http://doi.org/dbgw76. [ Links ]
16. Kaufmann SG, Westenbroek RE, Zechner C, Maass AH, Bischoff S, Muck J, et al. Functional protein expression of multiple sodium channel alpha- and beta-subunit isoforms in neonatal cardiomyocytes. J Mol Cell Cardiol. 2010; 48:261-9. http://doi.org/frbf4w. [ Links ]
17. Abriel H. Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2010; 48:2-11. http://doi.org/dmgrrt. [ Links ]
18. Herfst LJ, Rook MB, Jongsma HJ. Trafficking and functional expression of cardiac Na+ channels. J Mol Cell Cardiol. 2004; 36:185-93. http://doi.org/cbtrf2. [ Links ]
19. Abriel H. Cardiac sodium channel Nav1.5 and its associated proteins. Arch Mal Coeur Vaiss. 2007; 100:787-93. [ Links ]
20. Ashpole N, Herren A, Ginsburg K. Ca/Calmodulin-Dependent Protein Kinase II Regulates Cardic Sodium Channel Nav1.5 Gating by Multiple Phosphorylation Sites. J Biol Chem. 2012; 287:19856-69. http://doi.org/zd6. [ Links ]
21. Arora S, Mahajan B, Gupta K, Chopra P. Voltage-Gated Sodium Channels: Physiology, Pathology and Therapeutic Potential. Journal of Anaesthesiology Clinical Pharmacology. 2005; 21:125-36. [ Links ]
22. Gutiérrez DA, Fernández-Tenorio M, Ogrodnik J, Niggli E. NO-dependent CaMKII activation during beta-adrenergic stimulation of cardiac muscle. Cardiovasc Res. 2013; 100:392-401. http://doi.org/zd7. [ Links ]
23. Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol. 2010; 588:4969-85. http://doi.org/bx26dg. [ Links ]
24. Chatelier A, Mercier A, Tremblier B, Thériault O, Moubarak M, Benamer N, et al. A distinct de novo expression of Nav1.5 sodium channels in human atrial fibroblasts differentiated ito myofibroblasts. J Physiol. 2012; 590:4307-19. http://doi.org/zd8. [ Links ]
25. Osadchii OE, Soltysinska E, Olesen SP. Na+ channel distribution and electrophysiological heterogeneities in guinea pig ventricular wall. Am J Physiol Heart Circ Physiol. 2011; 300:H989-1002. http://doi.org/dvpfhd. [ Links ]
26. Beyder A, Strege PR, Reyes S, Bernard CE, Terzic A, Makielski J, et al. Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel Na(v)1.5: a novel mechanism of drug action. Circulation. 2012; 125:2698-706. http://doi.org/zd9. [ Links ]
27. Albesa M, Ogrodnik J, Rougier JS, Abriel H. Regulation of the cardiac sodium channel Nav1.5 by utrophin in dystrophin-deficient mice. Cardiovasc Res. 2011; 89:320-8. http://doi.org/bkhq2x. [ Links ]
28. Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, et al. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006; 99:407-14. http://doi.org/d5df5q. [ Links ]
29. Casini S, Verkerk AO, van Borren MM, van Ginneken AC, Veldkamp MW, de Bakker JM, et al. Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc Res. 2009; 81:72-81. http://doi.org/c2xr55. [ Links ]
30. Chagot B, Chazin WJ. Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J Mol Biol. 2011; 406:106-19. http://doi.org/cfmtgp. [ Links ]
31. Biswas S, DiSilvestre D, Tian Y, Halperin VL, Tomaselli GF. Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ Res. 2009; 104:870-8. http://doi.org/fjrk88. [ Links ]
32. Mario D. Connexin43 regulates sodium current; ankyrin-G modulates gap junctions: the intercalated disc exchanger. Cardiovasc Res. 2012; 93:220-2. http://doi.org/fbzgvn. [ Links ]
33. Casini S, Tan HL, Demirayak I, Remme CA, Amin AS, Scicluna BP, et al. Tubulin polymerization modifies cardiac sodium channel expression and gating. Cardiovasc Res. 2010; 85:691-700. http://doi.org/dbcztk. [ Links ]
34. Liu M, Liu H, Dudley SC. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010; 107:967-74. http://doi.org/crmdsb. [ Links ]
35. Johannessen M, Ramachandran S, Riemer L, Ramos-Serrano A, Ruoho AE, Jackson MB. Voltage-gated sodium channel modulation by sigma-receptors in cardiac myocytes and heterologous systems. Am J Physiol Cell Physiol. 2009; 296:C1049-57. http://doi.org/cjkr2z. [ Links ]
36. Wang DW, Mistry AM, Kahlig KM, Kearney JA, Xiang J, George AL. Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Front Pharmacol. 2010; 1:144. http://doi.org/bwvxz7. [ Links ]
37. Song W, Shou W. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr Cardiol. 2012; 33:943-9. http://doi.org/zfb. [ Links ]
38. Calloe K, Refaat MM, Grubb S, Wojciak J, Campagna J, Thomsen NM, et al. Characterization and mechanisms of action of novel NaV1.5 channel mutations associated with Brugada syndrome. Circ Arrhythm Electrophysiol. 2013; 6:177-84. http://doi.org/zfc. [ Links ]
39. Benito B, Brugada J, Brugada R, Brugada P. Síndrome de Brugada. Rev Esp Cardiol. 2009; 62:1297-315. http://doi.org/d5j8m2. [ Links ]
40. Muggenthaler M, Behr ER. Brugada syndrome and atrial fibrillation: pathophysiology and genetics. Europace. 2011; 13:913-5. http://doi.org/dmh8z4. [ Links ]
41. Chen L, Zhang W, Fang C, Jiang S, Shu C, Cheng H, et al. Polymorphism H558R in the human cardiac sodium channel SCN5A gene is associated with atrial fibrillation. J Int Med Res. 2011; 39:1908-16. http://doi.org/zfd. [ Links ]
42. Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation. 2008; 117:1927-35. http://doi.org/cdfb74. [ Links ]
43. Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005; 293:447-54. http://doi.org/bsjtjj. [ Links ]
44. Chen LY, Ballew JD, Herron KJ, Rodeheffer RJ, Olson TM. A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clin Pharmacol Ther. 2007;81:35-41. http://doi.org/b7x2mb. [ Links ]
45. Watanabe H, Darbar D, Kaiser DW, Jiramongkolchai K, Chopra S, Donahue BS, et al. Mutations in sodium channel beta1- and beta2-subunits associated with atrial fibrillation. Circ Arrhythm Electrophysiol. 2009; 2:268-75. http://doi.org/dfnfpc. [ Links ]
46. Yu J, Hu J, Dai X, Cao Q, Xiong Q, Liu X, et al. SCN5A mutation in Chinese patients with arrhythmogenic right ventricular dysplasia. Herz. 2014; 39:271-5. http://doi.org/zff. [ Links ]
47. Gillet L, Shy D, Abriel H. NaV1.5 and interacting proteins in human arrhythmogenic cardiomyopathy. Future Cardiol. 2013; 9:467-70. http://doi.org/zfg. [ Links ]
48. Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008; 18:78-87. http://doi.org/bbspqs. [ Links ]
49. Olesen MS, Jespersen T, Nielsen JB, Liang B, Moller DV, Hedley P, et al. Mutations in sodium channel beta-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc Res. 2011; 89:786-93. http://doi.org/c5vvpv. [ Links ]
50. Benito B, Brugada R, Perich RM, Lizotte E, Cinca J, Mont L, et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm. 2008; 5:1434-40. http://doi.org/fshq6v. [ Links ]
51. Gao G, Xie A, Huang SC, Zhou A, Zhang J, Herman AM, et al. Role of RBM25/LUC7L3 in abnormal cardiac sodium channel splicing regulation in human heart failure. Circulation. 2011; 124:1124-31. http://doi.org/c2kf8t. [ Links ]
52. Jones DK, Peters CH, Tolhurst SA, Claydon TW, Ruben PC. Extracellular proton modulation of the cardiac voltage-gated sodium channel, Nav1.5. Biophys J. 2011; 101:2147-56. http://doi.org/d3k3bz. [ Links ]
53. Marsman RF, Bezzina CR, Freiberg F, Verkerk AO, Adriaens ME, Podliesna S, et al. Coxsackie and adenovirus receptor (CAR) is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. J Am Coll Cardiol. 2014; 63:549-59. http://doi.org/f2qq5z. [ Links ]
