










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina
Print version ISSN 0120-0011
rev.fac.med. vol.63 no.1 Bogotá Jan./Mar. 2015
https://doi.org/10.15446/revfacmed.v63n1.45007
DOI: http://dx.doi.org/10.15446/revfacmed.v63n1.45007
CASOS CLÍNICOS
Síndrome de Alport: reporte de caso y revisión
Alport sindrome: Case report and literature review
José Augusto Urrego-Díaz1, Guillermo Landinez-Millán1, Carlos Javier Lozano-Triana1,2
1 Departamento de Pediatría, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá, D. C., Colombia.
2 Unidad de Medicina Interna, Hospital de la Misericordia. Bogotá, D. C., Colombia.
Correspondencia: José Augusto Urrego-Díaz. Carrera 14B No. 163-24 apto. 805, Tel: +57 1 8103204, Bogotá, D. C., Colombia. E-mail: joaurregodi@unal.edu.co
Recibido: 17/08/2014 Aceptado: 29/01/2015
Resumen
Se presenta el caso de una paciente de 4 años de edad, con hermano gemelo dicigoto asintomático, hija de padres no consanguíneos y sin antecedentes familiares de enfermedad renal. Inicia su cuadro clínico con edemas y proteinuria severa como manifestación de un síndrome nefrótico primario de cambios mínimos; este se diagnosticó por biopsia renal y, en un principio, se manejó con esteroides. Su evolución no fue adecuada debido a múltiples recaídas que la clasificaron como síndrome nefrótico corticorresistente. Por ello, se requirió un cambio en su tratamiento y una segunda biopsia renal, cuyo resultado histológico sorprendió al grupo médico tratante porque los cambios en la membrana basal glomerular confirmaban que se trataba de un Síndrome de Alport.
Palabras clave: Nefritis Hereditaria; Hematuria; Proteinuria; Síndrome de Alport (DeCS).
Urrego-Díaz JA, Landinez-Millán G, Lozano-Triana CJ. Síndrome de Alport: reporte de caso y revisión. Rev. Fac. Med. 2015;63(1):143-9. Spanish. doi: http://dx.doi.org/10.15446/revfacmed.v63n1.45007.
Summary
We present the case of a 4 year-old girl patient, with an asymptomatic dizygotic twin brother, child of non-consanguineous parents and with no family history of renal disease. Her clinical picture started with edema and severe proteinuria as manifestations of a minimal change nephrotic syndrome that was diagnosed by renal biopsy and initially treated with steroids. Her clinical course was complicated by multiple relapses that classified her as a patient presenting a steroid-resistant nephrotic syndrome, her treatment was changed and a second renal biopsy was needed. Histology outcome of biopsy surprised the treating medical group because changes in glomerular basal membrane revealed that it was in fact an Alport syndrome.
Keywords: Nephritis, hereditary; Hematuria; Proteinuria; Alport Syndrome (MeSH).
Urrego-Díaz JA, Landinez-Millán G, Lozano-Triana CJ. [Alport syndrome: Case report and literature review]. Rev. Fac. Med. 2015;63(1):143-9. Spanish. doi: http://dx.doi.org/10.15446/revfacmed.v63n1.45007.
Introducción
El síndrome de Alport (SA) fue descrito por primera vez por Cecil Alport en 1927 como una "nefritis hemorrágica hereditaria familiar congénita" (1). Esta patología es producto de una alteración de las membranas basales, secundaria a un defecto en el colágeno tipo IV que usualmente lleva a compromiso de la membrana basal glomerular con hematuria. Además, suele afectar el ojo y la cóclea, en diferentes magnitudes, dentro de una amplia variedad de fenotipos (2,3). El SA se encuentra en alrededor del 3 % de los niños con enfermedad renal terminal y en un 0,2% de los adultos en Estados Unidos (2), con una incidencia y severidad mayor en hombres que en mujeres (4).
No existe casuística similar de este síndrome en Colombia. Hace más de tres décadas se demostró la naturaleza genética heterogénea de esta enfermedad: se presenta principalmente en su forma ligada al cromosoma X y, en menor medida, en sus formas autosómica recesiva y autosómica dominante (5,6). La forma ligada al cromosoma X representa entre el 80 y 85% de los casos y se debe a una mutación en el gen COL4A5 (7). La forma autosómica recesiva está presente en el 15% de las familias y se da por ciertas mutaciones en los genes COL4A3 o COL4A4, ubicados en el cromosoma 2q35-37 (8,9); mientras que la forma autosómica dominante se presenta en el 5% de las familias y es causada por otras mutaciones también en los genes COL4A3 o COL4A4 (9,10).
A continuación, se mostrará el caso de una paciente que se presentó, en principio, con manifestaciones clínicas de síndrome nefrótico y que eventualmente terminó con el diagnóstico de SA.
Presentación del caso
Al Hospital de la Misericordia (HOMI) de Bogotá (Colombia), llegó una paciente de 3 años de edad, con antecedente de síndrome nefrótico en estudio por nefrología pediátrica de forma ambulatoria. Dicho síndrome inició a los 2 años de edad, luego de un cuadro de síntomas respiratorios altos y fiebre; y se caracterizó por edemas, orina hipercoloreada y espumosa, con presencia en el examen general de orina de hematuria y proteinuria en rango no nefrótico. En controles posteriores, la proteinuria aumentó hasta rango nefrótico y la hematuria disminuyó en intensidad progresivamente sin manejo farmacológico.
Algunos exámenes de laboratorio realizados en consulta externa de nefrología pediátrica fueron: complemento C3 y C4 normales, serología para hepatitis B y C, rubeola, citomegalovirus, herpes 1 y 2, VIH y Epstein Barr negativa, y P-ANCAS, C-ANCAS y anticuerpos anti-membrana basal glomerular negativos. 6 meses después del inicio de los estudios, se realizó biopsia renal, cuyo resultado fue esclerosis focal sin cambios en la célula endotelial. No se realizó microscopía electrónica ni inmunofluorescencia debido a que la muestra fue insuficiente; por ende, se decidió realizar una nueva biopsia renal, que fue tomada 6 meses después y cuyo reporte quedó pendiente. En los antecedentes farmacológicos, se encontró que la paciente llevaba 6 semanas de tratamiento con prednisolona, a dosis de 60mg/m2/día, debido a la persistencia de proteinuria en rango nefrótico. Hasta entonces, no había presentado cifras tensionales elevadas.
La paciente ingresó luego al servicio de urgencias en estatus convulsivo sin antecedentes de trauma, epilepsia o fiebre. La paciente es fruto de la segunda gestación de un embarazo gemelar dicigótico con padres no consanguíneos. Al ingreso, se encontraron cifras tensionales elevadas de 148/98, glucometría de 82, sin alteración hidroelectrolítica. Llamó la atención su evidente fascies cushinoide. La paciente fue manejada con benzodiacepinas, cediendo el cuadro convulsivo con posterior recuperación del estado de conciencia sin evidencia de signos de focalización. Se tomó una TAC de cráneo simple que no evidenció lesión estructural. Por la persistencia de cifras tensionales muy elevadas, se consideró que estaba cursando con una emergencia hipertensiva con órgano blanco en el sistema nervioso central. Debido a esto, fue trasladada a la unidad de cuidados intensivos pediátricos (UCIP) para continuar su manejo integral y monitoreo. En la UCIP fue manejada por 3 días con múltiples antihipertensivos orales y endovenosos, con los que se controló la crisis hipertensiva, pero con persistencia de cifras tensionales elevadas. Durante este tiempo, llamó la atención la proteinuria en rango nefrótico, a pesar de 6 semanas de manejo con corticoide. Por lo anterior, se consideró que la paciente cursaba un síndrome nefrótico corticorresistente (SNCR). El servicio de nefrología pediátrica inició manejo para SNCR con protocolo de Mendoza, aplicando bolos de metilprednisolona por 5 días y uno de ciclofosfamida con MESNA. No hubo mejoría de la proteinuria y, debido a la persistencia de hematuria microscópica e hipertensión arterial de difícil manejo, se clasificó como una glomerulopatía compleja. Por lo tanto se intentó iniciar manejo con micofenolato, que no fue autorizado por su EPS; por esta razón, se optó por iniciar tratamiento con azatriopina y descenso progresivo del corticoide.
Durante su manejo en el servicio de Medicina Interna, una semana tras su egreso de la UCIP, se recibió el reporte de la segunda biopsia renal, que informó: inmunofluorescencia directa negativa para inmunocomplejos, ultraestructura con membranas muy irregulares con aspecto de "piel de lagarto" y alteración de la lámina densa con ruptura y lamelación. Conclusión: cambios compatibles con anomalía estructural del colágeno de la membrana basal capilar de tipo nefritis hereditaria/síndrome de Alport. Se realizó valoración por oftalmología, que no reportó anormalidades, y potenciales auditivos evocados, que se encontraron dentro de límites normales. Fue finalmente valorada por el área de Genética; allí se iniciaron los estudios respectivos, incluyendo la familia, principalmente al hermano gemelo, quien fue encontrado asintomático. La paciente fue dada de alta con dosis interdiarias de corticoide en descenso, azatriopina y dosis plenas de antihipertensivos orales.
Síndrome de Alport
El síndrome de Alport (SA) es un conjunto de enfermedades que se caracterizan por afección hereditaria de la membrana basal glomerular por alteraciones en el colágeno tipo IV que la compone. Se presenta con hematuria micro- o macroscópica; además, suele asociarse a alteraciones auditivas y oculares, y es causa de alrededor de 0.3 a 3% de la enfermedad renal terminal en pediatría (2,11,12).
El colágeno tipo IV es una familia de 6 proteínas, las cadenas α1(IV) a α6(IV), cada una codificada por su propio gen (COL4A1 a COL4A6) (13). Los genes COL4A1 y COL4A2 se encuentran en el cromosoma 13 (14), COL4A3 y COL4A4 se encuentran en el cromosoma 2 (15), y COL4A5 y COL4A6 están en el cromosoma X (16,17). El SA es causado por mutaciones en los genes que codifican para las cadenas α3(IV), α4(IV) o α5(IV) del colágeno tipo IV (13), isoformas que van prevaleciendo sobre las otras en el riñón, a medida que el niño crece. Esto explica la naturaleza progresiva de la enfermedad.
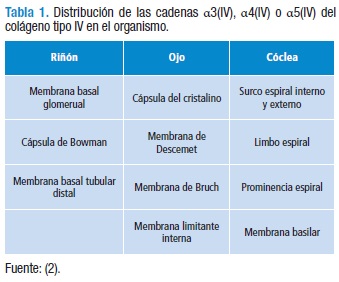
Distintas isoformas del colágeno tipo IV están presentes en diferentes membranas y diferentes órganos (2). La distribución de las cadenas α3(IV), α4(IV) o α5(IV) en el organismo (tabla 1) explica el compromiso de riñones, ojos y cóclea en el paciente con SA.
Según el patrón de herencia genética, el SA se ha clasificado en 3 categorías:
Ligado al cromosoma X (SALX): Se da por la mutación del gen COL4A5, que codifica para la cadena α5 y está ubicado en el brazo largo del cromosoma Xq22-24 (2). Esta mutación es la más frecuente, pues se encuentra en un 80 a 85% de los casos (7, 18). Cerca del 10 al 15% de las mutaciones se dan de novo, y suelen ocurrir en el gameto de un padre (19). Lo portan ambos sexos, pero son los varones quienes desarrollan la patología con mayor gravedad hasta la enfermedad renal terminal; mientras que las mujeres heterocigotas solo suelen presentar episodios esporádicos de hematuria o, incluso, permanecen toda su vida sin manifestaciones (13).
Autosómico recesivo (SAAR): Por mutaciones en los genes COL4A3 o COL4A4, que codifican para las cadenas α3 y α4, respectivamente, y están ubicados en el cromosoma 2q35-37 (20, 21). Esta mutación tiene una prevalencia de 15% en los casos de SA (7) y afecta con una severidad similar a hombres y mujeres (22). La mutación heterocigota produce cuando mucho hematuria asintomática, pero la unión de dos individuos portadores resultará en un niño con ARAS (13). Por lo tanto en estos casos, como en todas las enfermedades autosómicas recesivas, es importante el antecedente de consanguinidad entre los padres.
Autosómico dominante (SAAD): Producto de mutaciones en los genes COL4A3 y COL4A4 que en su forma heterocigota producen un SA claramente definido (13). Es la que se presenta con menor prevalencia, siendo alrededor de 5% de los casos de SA (7), aunque un estudio reciente (en el que se usaron nuevas tecnologías de secuenciación) llamó la atención al encontrar una prevalencia de 30% (23).
Los síndromes de Epstein y Fechtner se caracterizan por nefritis progresiva y macrotrombocitopenia de herencia autosómica dominante, que, si bien en ocasiones en la literatura se confunden con una variante del SAAD, son totalmente diferentes. Se producen por mutaciones en la cadena pesada de miosina no muscular IIA (13,24).
Si bien con frecuencia se encuentra historia familiar de hematuria en los pacientes con SA, este síndrome puede ocurrir esporádicamente como resultado de mutaciones de novo (13).
Las manifestaciones clínicas del SA son:
Nefropatía: La hematuria microscópica es la manifestación que acompaña siempre a los pacientes con síndrome de Alport, quienes también pueden presentar episodios de hematuria macroscópica, precipitados generalmente por infecciones respiratorias superiores (7, 13). Las mujeres heterocigotas para SALX suelen presentar episodios intermitentes de hematuria o, incluso, en un 10% de los casos, jamás presentarla; en contraste, tanto hombres como mujeres afectados por SAAR presentan hematuria de forma persistente (2).
La proteinuria suele estar ausente en los primeros años, pero se desarrollará finalmente en los hombres afectados por SALX y en ambos sexos en el caso de SAAR (2). En raras ocasiones una mujer heterocigota para SALX presenta proteinuria significativa (2). Lo usual es encontrar la proteinuria de forma progresiva, aumentando con la edad hasta un franco síndrome nefrótico, siendo entonces muy inusual el debut del síndrome de Alport a través de un síndrome nefrótico (13).
La hipertensión se presenta de modo similar a la proteinuria: aumenta su prevalencia y severidad con la edad, es más frecuente su desarrollo en hombres que en mujeres afectados por SALX y no presenta diferencias en prevalencia entre ellos y ellas en el SAAR (13).
Defectos auditivos: Hasta el 90% de los hombres con SALX presentan pérdida de la audición sensorineural a los 40 años (25). La evaluación audiológica suele mostrar su inicio finalizando la niñez, y en algunos casos, principalmente en mujeres, puede presentarse hasta edades tardías de la vida o incluso nunca presentarse (3, 13).
Todos los pacientes con la enfermedad recesiva presentarán sordera, sin importar el sexo (13). El defecto es, en principio, para tonos agudos pero va progresando a otras frecuencias (7). Estos pacientes no tienen una sordera congénita; por el contrario, la afección es progresiva y aumenta en prevalencia y en nivel de compromiso con la edad, motivo por el cual los exámenes auditivos deben ser periódicos.
La sordera en el síndrome de Alport se debe a la pérdida de la red del colágeno α3(IV)- α4(IV)- α5(IV) en las membranas basales de la cóclea (26).
Patología oftalmológica: Existe un signo clínico distintivo, que es patognomónico del síndrome de Alport: el lenticono anterior, que se caracteriza por la protrusión hacia la cámara anterior de una protuberancia del centro del cristalino (2). Todos los pacientes en quienes se evidencia lenticono anterior presentan también algún grado de nefritis crónica y de sordera sensorineural y antes de los 30 años habrán progresado a enfermedad renal terminal y sordera (13, 27). Esta afección es mucho más frecuente en hombres que en mujeres, no está presente al nacimiento y aparece usualmente durante la segunda o tercera década de la vida (13).
Otra manifestación que se presenta hasta en el 14% de los pacientes con SA es una maculopatía consistente en manchas blanquecinas o amarillentas perimaculares, que son –al parecer– asintomáticas (28). También pueden presentarse distrofia corneal posterior, por defectos en la membrana de Descemet (la membrana basal del endotelio corneal) (29,30), y erosiones corneales recurrentes, debido a alteraciones de la membrana basal epitelial corneal (31,32).
Leiomiomatosis: En algunos pacientes con SALX puede presentarse leiomiomatosis en diferentes zonas: En el esófago, que puede manifestarse con dolor retroesteronal o epigástrico, disfagia y vómito posprandial; en el árbol traqueobronquial, que se manifiesta con bronquitis recurrente, tos, disnea y estridores, y en el tracto genital femenino (33-35).
El diagnóstico de SA en un paciente en quien se sospeche puede confirmarse a través de biopsias de riñón o de piel o con pruebas genéticas (7).
Dado que el colágeno tipo IV también se expresa en la piel y que las biopsias de piel son menos invasivas que las de riñón, estas biopsias deberían ser la primera aproximación en caso de sospecha de SA (2). Alrededor del 80% de los hombres y 60% de las mujeres con SALX no tendrán cadenas α5(IV) del colágeno en la membrana basal epidérmica, siendo en hombres total la pérdida de esta cadena y en mujeres una pérdida segmentaria (36,37). Además, existe evidencia de que muchos individuos con SALX tienen anormalidades en la expresión de las cadenas α2(IV) en la piel y que la mayoría de individuos con SAAR no expresan las cadenas α3(IV), α4(IV) o α5(IV) en este órgano (38).
En caso de no confirmarse el diagnóstico a través de una biopsia de piel se indicaría la biopsia renal (2). En este caso, las cadenas α3(IV), α4(IV) o α5(IV) en la mayoría de glomérulos, túbulos y cápsulas de Bowman serían indetectables por inmunohistoquímica en un paciente masculino afectado por SALX; mientras que en una mujer habría compromisos segmentarios de la membrana basal glomerular (39). Para el caso de SAAR no se identificarían las cadenas α3(IV) o α4(IV) en la membrana basal glomerular o membrana basal tubular (40).
Además, existe toda una serie de hallazgos ultraestructurales característicos en la membrana basal glomerular y la lámina densa en el SA, evidenciables a través de microscopía electrónica en biopsias de piel o riñones (2,7).
Finalmente, si el diagnóstico todavía es incierto pueden realizarse pruebas genéticas (2). La detección de mutaciones de COL4A3, COL4A4 y COL4A5 es en extremo difícil y costosa, se hace en pocos laboratorios y consume un tiempo considerable, razón por la cual debería realizarse solo cuando existe duda sobre el diagnóstico tras las biopsias o cuando se desea realizar un diagnóstico prenatal (41,42).
No existen ensayos clínicos aleatorizados que ofrezcan información sobre el enfoque ideal del tratamiento del SA; por ello, éste se basa en la experiencia clínica, pequeños estudios y opinión de expertos (7,13). El manejo de esta entidad va encaminado a frenar su progresión y en forma final al trasplante renal como intervención definitiva en la falla renal.
En ausencia de datos, el uso de IECA y ARAII parece ser un tratamiento aceptable en pacientes con SA que comienzan a presentar proteinuria, pues disminuyen esta manifestación (43). Además, algunos estudios han reportado que la combinación de inhibidores de aldosterona con estos medicamentos puede aumentar su control sobre la proteinuria en niños con SA (44,45). Asímismo, se ha demostrado que la ciclosporina es útil para disminuir la proteinuria en pacientes y animales con síndrome nefrótico corticorresistente (46-48), como el caso aquí presentado; no obstante, debe evitarse su uso prolongado por la posibilidad de nefrotoxicidad (48).
El trasplante renal sigue siendo el único tratamiento definitivo en el SA, con buena tasa de supervivencia del injerto en general (43). Se debe tener presente que los pacientes que reciben este tratamiento tienen la posibilidad de desarrollar glomerulonefritis anti-BMG, posterior al procedimiento. Ésta es una complicación infrecuente pero catastrófica, que se desarrolla por producción de anticuerpos contra las cadenas de colágeno del injerto, para las cuales el huésped no ha establecido tolerancia inmune (49). Esta complicación afecta a alrededor de 2-3% de los pacientes del sexo masculino con SALX y, en menor porcentaje, a mujeres o a pacientes con progresión lenta a enfermedad renal terminal (13,50).
Discusión
Tras revisar los registros del HOMI, se encontró que tan solo se contaba con 4 casos de SA confirmados en los últimos años. Aunque la frecuencia del SA es relativamente baja (causa del 3% de los casos de enfermedad renal terminal en niños) (2), se esperaría encontrar un mayor número de casos. Esta baja cantidad puede deberse a que en algunas ocasiones el SA no se sospecha y por lo tanto no se realizan los métodos (mencionados en la anterior sección) que permitan confirmar su diagnóstico, de esta forma, un paciente puede terminar con un tratamiento distinto bajo el diagnóstico de algún otro tipo de nefritis.
Algunas características presentadas en el cuadro de la paciente llaman la atención y merecen ser analizadas a la luz del conocimiento actual sobre el SA.
El hecho de que la paciente reportada no tuviera antecedentes familiares de SA y, aún más, que su propio hermano gemelo dicigótico no presentara manifestaciones clínicas ni paraclínicas de esta entidad en algún momento puede hacer poner en duda el diagnóstico de una enfermedad netamente hereditaria, como es el SA. Sin embargo, la literatura demuestra que alrededor del 10 al 15% de las mutaciones en el SALX son de novo, en el gameto de un padre (19).
Si bien es poco probable que el caso de esta paciente sea un SALX, ya que esta variante no suele afectar con tal intensidad al sexo femenino (13), sí puede ser una de las otras dos variantes hereditarias de esta enfermedad, que afectan con similar intensidad a ambos sexos (22). Asimismo, que la causante del SA en esta paciente fuera una mutación de novo en alguna de las líneas germinales de alguno o ambos padres explicaría la ausencia de esta enfermedad en su gemelo. Este, al ser dicigótico, podría tener una carga genética tan diferente a la de ella como la de cualquier hermano no gemelar.
Luego de las valoraciones audiológica y oftalmológica a la paciente se le descartó compromiso auditivo y ocular, lo que es usual en los primeros años de vida en el SA; esto se debe a que dichos cambios se desarrollan progresivamente, evidenciándose entre el final de la niñez y la adultez (3,13,25). Por lo anterior, en los casos de SA se requiere de vigilancia clínica a través de valoraciones que se realicen de manera periódica como está sucesiendo con la paciente.
La manifestación inicial del SA en la paciente fue hematuria macroscópica precipitada por una infección de vías respiratorias superiores, lo que es bastante usual en la literatura sobre el SA (7,13). Sin embargo, un aspecto curioso del cuadro de esta paciente fue la rápida evolución a síndrome nefrótico, desde su debut se presentó con proteinuria significativa, que en cuestión de días evolucionó a proteinuria en rango nefrótico. Esto es realmente inusual, pues el SA no suele debutar con proteinuria significativa; ésta suele estar ausente en los primeros años y, por el contrario, evoluciona de forma lenta y progresiva, a medida que se instaura la falla renal (2,13). Vale la pena informar esta presentación, pues futuros estudios y reportes de casos podrían arrojar luz sobre esta clínica atípica e inexplorada del SA.
El diagnóstico de esta paciente se realizó por medio de una biopsia de riñón como es recomendado en la literatura (7), si bien se pasó por alto la realización previa de una biopsia de piel, que también podría haber hecho el diagnóstico y es de naturaleza menos invasiva que la biopsia renal (2). Esto es comprensible si se tiene en cuenta el cuadro clínico atípico que presentó, que no generó sospecha de SA; por el contrario, fue una sorpresa el resultado de la biopsia.
Finalmente, el pronóstico de esta paciente no es muy bueno. El tratamiento médico que se le dio y que se usa con frecuencia en el SA intenta ralentizar la evolución de la falla renal (43-48); no obstante, eventualmente necesitará un trasplante renal como tratamiento definitivo (43).
Conclusiones
Típicamente el SA se manifiesta, en principio, a través de hematuria microscópica persistente, con episodios de hematuria macroscópica y un desarrollo progresivo a hipertensión arterial, proteinuria y síndrome nefrótico. Sin embargo, el caso que aquí se presenta podría ser de los primeros que reporta presentaciones atípicas de este síndrome; se espera que futuros reportes arrojen luces sobre estas presentaciones.
El diagnóstico del SA se debe confirmar a través de biopsias de piel, renales o de pruebas genéticas; este es el orden que debe seguirse cuando se sospeche esta entidad. Por otro lado, se debe realizar una adecuada observación a los familiares de los pacientes con SA confirmado y evaluar la realización de un estudio genético, si bien hasta el momento dicho estudio no está estandarizado.
Conflicto de intereses
Ninguno declarado por los autores.
Financiación
Ninguna declarada por los autores.
Agradecimientos
Ninguno declarado por los autores
Referencias
1. Alport AC. Hereditary familial congenital haemorrhagic nephritis. Br Med J. 1927 [cited 2015 feb 12];1(3454):504-6. doi: http://doi.org/cqx6nk. [ Links ]
2. Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine (Baltimore). 1999 [cited 2015 feb 12];78(5):338-60. doi: http://doi.org/fmtssz. [ Links ]
3. Barker DF, Pruchno CJ, Jiang X, Atkin CL, Stone EM, Denison JC, et al. A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am J Hum Genet. 1996;58(6):1157-65. [ Links ]
4. Gretz N, Broyer M, Brunner FP, Brynger H, Donckerwolcke RA, Jacobs C, et al. Alport's syndrome as a cause of renal failure in Europe. Pediatr Nephrol. 1987 [cited 2015 feb 12];1(3):411-5. doi: http://doi.org/ffd594. [ Links ]
5. Feingold J, Bois E, Chompret A, Broyer M, Gubler MC, Grünfeld JP. Genetic heterogeneity of Alport syndrome. Kidney Int. 1985 [cited 2015 feb 12];27(4):672-7. doi: http://doi.org/dq4m7n. [ Links ]
6. O'Neill WM, Atkin CL, Bloomer HA. Hereditary nephritis: a re-examination of its clinical and genetic features. Ann Intern Med. 1978 [cited 2015 feb 12];88(2):176-82. doi: http://doi.org/zr8. [ Links ]
7. Medeiros M, Fuentes Y, García P, Hernández A, Morán F, Velásquez L. Síndrome de Alport. Bol Med Hosp Infant Mex. 2008;65:331-40. [ Links ]
8. Heidet L, Arrondel C, Forestier L, Cohen-Solal L, Mollet G, Gutierrez B, et al. Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol. 2001;12(1):97-106. [ Links ]
9. Longo I, Porcedda P, Mari F, Giachino D, Meloni I, Deplano C, et al. COL4A3/COL4A4 mutations: from familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002 [cited 2015 feb 12];61(6):1947-56. doi: http://doi.org/fr8x7z. [ Links ]
10. Slajpah M, Gorinsek B, Berginc G, Vizjak A, Ferluga D, Hvala A, et al. Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int. 2007 [cited 2015 feb 12];71(12):1287-95. doi: http://doi.org/d3skt5. [ Links ]
11. Rheault MN. Women and Alport syndrome. Pediatr Nephrol. 2012 [cited 2015 feb 12];27(1):41-6. doi: http://doi.org/dsmf5p. [ Links ]
12. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol. 2010 [cited 2015 feb 12];21(5):876-83. doi: http://doi.org/bpf3x6. [ Links ]
13. Kashtan CE. Alport Syndrome and Thin Basement Membrane Nephropathy: Diseases Arising from Mutations in Type IV Collagen. Saudi J Kidney Dis Transpl. 2003 [cited 2015 feb 12];14(3):276-89. [ Links ]
14. Boyd CD, Toth-Fejel SE, Gadi IK, Litt M, Condon MR, Kolbe M, et al. The genes coding for human pro alpha 1(IV) collagen and pro alpha 2(IV) collagen are both located at the end of the long arm of chromosome 13. Am J Hum Genet. 1988 [cited 2015 feb 12];42(2):309-14. [ Links ]
15. Mariyama M, Zheng K, Yang-Feng TL, Reeders ST. Colocalization of the genes for the alpha 3(IV) and alpha 4(IV) chains of type IV collagen to chromosome 2 bands q35-q37. Genomics. 1992 [cited 2015 feb 12];13(3):809-13. doi: http://doi.org/dpb5rx. [ Links ]
16. Hostikka SL, Eddy RL, Byers MG, Höyhtyä M, Shows TB, Tryggvason K. Identification of a distinct type IV collagen alpha chain with restricted kidney distribution and assignment of its gene to the locus of X chromosome-linked Alport syndrome. Proc Natl Acad Sci U S A. 1990 [cited 2015 feb 12];87(4):1606-10. doi: http://doi.org/fxhqxh. [ Links ]
17. Myers JC, Jones TA, Pohjolainen ER, Kadri AS, Goddard AD, Sheer D, et al. Molecular cloning of alpha 5(IV) collagen and assignment of the gene to the region of the X chromosome containing the Alport syndrome locus. Am J Hum Genet. 1990 [cited 2015 feb 12];46(6):1024-33. [ Links ]
18. Kashtan CE. Familial hematuria due to type IV collagen mutations: Alport syndrome and thin basement membrane nephropathy. Curr Opin Pediatr. 2004 [cited 2015 feb 12];16(2):177-81. doi: http://doi.org/bnc79s. [ Links ]
19. Lemmink HH, Schröder CH, Monnens LA, Smeets HJ. The clinical spectrum of type IV collagen mutations. Hum Mutat. 1997 [cited 2015 feb 12];9(6):477-99. doi: http://doi.org/bz9rjs. [ Links ]
20. Longo I, Scala E, Mari F, Caselli R, Pescucci C, Mencarelli MA, et al. Autosomal recessive Alport syndrome: an in-depth clinical and molecular analysis of five families. Nephrol Dial Transplant. 2006 [cited 2015 feb 12];21(3):665-71. doi: http://doi.org/cdgr6m. [ Links ]
21. Uzak AS, Tokgoz B, Dundar M, Tekin M. A novel COL4A3 mutation causes autosomal-recessive Alport syndrome in a large Turkish family. Genet Test Mol Biomarkers. 2013 [cited 2015 feb 12];17(3):260-4. doi: http://doi.org/zr9. [ Links ]
22. Wang Y, Sivakumar V, Mohammad M, Colville D, Storey H, Flinter F, et al. Clinical and genetic features in autosomal recessive and X-linked Alport syndrome. Pediatr Nephrol. 2014 [cited 2015 feb 12];29(3):391-6. http://doi.org/zsb. [ Links ]
23. Fallerini C, Dosa L, Tita R, Del Prete D, Feriozzi S, Gai G, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet. 2014 [cited 2015 feb 12];86(3):252-7. doi: http://doi.org/zsc. [ Links ]
24. Heath KE, Campos-Barros A, Toren A, Rozenfeld-Granot G, Carlsson LE, Savige J, et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am J Hum Genet. 2001 [cited 2015 feb 12];69(5):1033-45. doi: http://doi.org/d5n2xx. [ Links ]
25. Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. 2000;11(4):649-57. [ Links ]
26. Kalluri R, Gattone VH, Hudson BG. Identification and localization of type IV collagen chains in the inner ear cochlea. Connect Tissue Res. 1998 [cited 2015 feb 12];37(1-2):143-50. doi: http://doi.org/ccgz4f. [ Links ]
27. Nielsen CE. Lenticonus anterior and Alport's syndrome. Acta Ophthalmol (Copenh). 1978 [cited 2015 feb 12];56(4):518-30. doi: http://doi.org/dgr5zp. [ Links ]
28. Perrin D, Jungers P, Grünfeld JP, Delons S, Noël LH, Zenatti C. Perimacular changes in Alport's syndrome. Clin Nephrol. 1980 [cited 2015 feb 12];13(4):163-7. [ Links ]
29. Teekhasaenee C, Nimmanit S, Wutthiphan S, Vareesangthip K, Laohapand T, Malasitr P, et al. Posterior polymorphous dystrophy and Alport syndrome. Ophthalmology. 1991 [cited 2015 feb 12];98(8):1207-15. doi: http://doi.org/zsd. [ Links ]
30. Thompson SM, Deady JP, Willshaw HE, White RH. Ocular signs in Alport's syndrome. Eye (Lond). 1987 [cited 2015 feb 12];1:146-53. doi: http://doi.org/c2brbt. [ Links ]
31. Burke JP, Clearkin LG, Talbot JF. Recurrent corneal epithelial erosions in Alport's syndrome. Acta Ophthalmol (Copenh). 1991 [cited 2015 feb 12];69(4):555-7. doi: http://doi.org/dhkghg. [ Links ]
32. Rhys C, Snyers B, Pirson Y. Recurrent corneal erosion associated with Alport's syndrome. Rapid communication. Kidney Int. 1997 [cited 2015 feb 12];52(1):208-11. doi: http://doi.org/dv7j4v. [ Links ]
33. Antignac C, Heidet L. Mutations in Alport syndrome associated with diffuse esophageal leiomyomatosis. Contrib Nephrol. 1996;117:172-82. [ Links ]
34. Mothes H, Heidet L, Arrondel C, Richter KK, Thiele M, Patzer L, et al. Alport syndrome associated with diffuse leiomyomatosis: COL4A5-COL4A6 deletion associated with a mild form of Alport nephropathy. Nephrol Dial Transplant. 2002 [cited 2015 feb 12];17(1):70-4. doi: http://doi.org/d73nhp. [ Links ]
35. Heidet L, Cohen-Solal L, Boye E, Thorner P, Kemper MJ, David A, et al. Novel COL4A5/COL4A6 deletions and further characterization of the diffuse leiomyomatosis-Alport syndrome (DL-AS) locus define the DL critical region. Cytogenet Cell Genet. 1997 [cited 2015 feb 12];78(3-4):240-6. doi: http://doi.org/dcktsm. [ Links ]
36. Rana K, Wang YY, Powell H, Jones C, McCredie D, Buzza M, et al. Persistent familial hematuria in children and the locus for thin basement membrane nephropathy. Pediatr Nephrol. 2005 [cited 2015 feb 12];20(12):1729-37. doi: http://doi.org/d9qg9w. [ Links ]
37. Massella L, Onetti Muda A, Faraggiana T, Bette C, Renieri A, Rizzoni G. Epidermal basement membrane alpha 5(IV) expression in females with Alport syndrome and severity of renal disease. Kidney Int. 2003 [cited 2015 feb 12];64(5):1787-91. doi: http://doi.org/dwksq9. [ Links ]
38. Patey-Mariaud de Serre N, Garfa M, Bessiéres B, Noël LH, Knebelmann B. Collagen alpha5 and alpha2(IV) chain coexpression: analysis of skin biopsies of Alport patients. Kidney Int. 2007 [cited 2015 feb 12];72(4):512-6. doi: http://doi.org/fxb9qt. [ Links ]
39. Kleppel MM, Santi PA, Cameron JD, Wieslander J, Michael AF. Human tissue distribution of novel basement membrane collagen. Am J Pathol. 1989;134(4):813-25. [ Links ]
40. Gubler MC, Knebelmann B, Beziau A, Broyer M, Pirson Y, Haddoum F, et al. Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int. 1995 [cited 2015 feb 12];47(4):1142-7. doi: http://doi.org/dvcb2z. [ Links ]
41. Gubler MC. Diagnosis of Alport syndrome without biopsy? Pediatr Nephrol. 2007 [cited 2015 feb 12];22(5):621-5. doi: http://doi.org/bqxwcv. [ Links ]
42. Artuso R, Fallerini C, Dosa L, Scionti F, Clementi M, Garosi G, et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur J Hum Genet. 2012 [cited 2015 feb 12];20(1):50-7. doi: http://doi.org/ftfdqm. [ Links ]
43. Kashtan CE. Familial hematuria. Pediatr Nephrol. 2009 [cited 2015 feb 12];24(10):1951-8. doi: http://doi.org/ft28vg. [ Links ]
44. Kaito H, Nozu K, Iijima K, Nakanishi K, Yoshiya K, Kanda K, et al. The effect of aldosterone blockade in patients with Alport syndrome. Pediatr Nephrol. 2006 [cited 2015 feb 12];21(12):1824-9. doi: http://doi.org/bqw8xp. [ Links ]
45. Giani M, Mastrangelo A, Villa R, Turolo S, Marra G, Tirelli AS, et al. Alport syndrome: the effects of spironolactone on proteinuria and urinary TGF-β1. Pediatr Nephrol. 2013 [cited 2015 feb 12];28(9):1837-42. doi: http://doi.org/zsf. [ Links ]
46. Desassis JF, Raats CJ, Bakker MA, van den Born J, Berden JH. Antiproteinuric effect of ciclosporin A in adriamycin nephropathy in rats. Nephron. 1997 [cited 2015 feb 12];75(3):336-41. doi: http://doi.org/b2j8z5. [ Links ]
47. Singh A, Tejani C, Tejani A. One-center experience with cyclosporine in refractory nephrotic syndrome in children. Pediatr Nephrol. 1999 [cited 2015 feb 12];13(1):26-32. doi: http://doi.org/cf85cg. [ Links ]
48. Charbit M, Gubler MC, Dechaux M, Gagnadoux MF, Grünfeld JP, Niaudet P. Cyclosporin therapy in patients with Alport syndrome. Pediatr Nephrol. 2007 [cited 2015 feb 12];22(1):57-63. doi: http://doi.org/c245bq. [ Links ]
49. McCoy RC, Johnson HK, Stone WJ, Wilson CB. Absence of nephritogenic GBM antigen(s) in some patients with hereditary nephritis. Kidney Int. 1982 [cited 2015 feb 12];21(4):642-52. doi: http://doi.org/fbv9v9. [ Links ]
50. Kashtan CE. Alport syndrome: renal transplantation and donor selection. Ren Fail. 2000 [cited 2015 feb 12];22(6):765-8. doi: http://doi.org/ct9xbh. [ Links ]
