











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Tumorigenesis is a complex multistage process which involves tumor initiation, promotion, progression to malignancy and metastasis. Tumor cells are characterized mainly by the result of uncontrolled proliferation processes, where cell division occurs faster. In addition to proliferation, other affected molecular mechanisms are programmed cell death or apoptosis, and the cell cycle 1.
Autophagy plays an important role, not only in the different stages of tumorigenesis, but also in disease states that lead to a microenvironment that promotes tumorigenesis. The role of this process in pathological states associated with higher risk of cancer, such as chronic liver disease, obesity and inflammatory bowel disease, is increasingly clear 2-4.
Pharmacological management of autophagy, with the intention of preventing a favorable microenvironment for tumor initiation, may require an opposite approach to limit tumor progression once pre-malignant cells are established. In this review, the regulation of autophagy, types of autophagy, autophagy itself and its mechanism as tumor suppressor or inducer are addressed. Finally, autophagy as a therapy against cancer, mainly in tumor cells with competent autophagy and defective autophagy, and induction of cell death by autophagy as a therapeutic strategy are discussed.
Molecular regulation of autophagy
Autophagy is a mechanism essential for maintaining cellular homeostasis in the body in the absence of important nutrients that work as an energy source. This process begins with the retention of cytoplasmic components, such as protein aggregates and damaged or aged organelles, through double-membrane vesicles called autophagosomes. The retentate is then transferred to degradation organelles such as lysosomes or vacuoles for destruction and eventual recycling of resulting macromolecules 5-7.
Although the study of the components of autophagy in mammalian cells was first performed in 1950, currently, it has been proved that there are studies performed in this population, and many others conducted using microorganisms such as yeast, where the existence of about 31 ATG genes has been observed; these genes are closely related to autophagy and the place where they are located in is known as perivacuolar site (PAS). The ongoing study of the nature of autophagy is becoming more important; here, the animal model in yeast is a powerful medium to decipher multiple concerns 8,9.
The molecular regulation of autophagy occurs in two different ways: a) through the activation of mTORC1 (mammalian Target of Rapamycin), in response to starvation or exhaustion of energy, and b) through energy detection, regulated by AMP-activated protein kinase (AMPK) 10. Autophagy could also be regulated by the Beclin 1 protein complex, consisting of the Beclin 1 (homolog Atg6) protein, phosphatidyl-inositol 3- kinase (PI3K) class III (PI3KC3/Vps34), p150, and Atg14L or UV radiation resistance-associated gene protein (UVRAG) 11,12. The intrinsic activation of the PI3KC3/Beclin 1 complex leads to the generation of phosphatidylinositol-3-phosphate (PI3P), which is required for the formation of the autophagosome. Moreover, there are several tumor suppressor proteins such as Atg4c, BAX-interacting factor-1 (Bif-1) homolog of phosphatase and tensin (PTEN) and UVRAG, which, besides inhibiting the growth of tumors, have common induction of autophagy 13 (Figure 1).
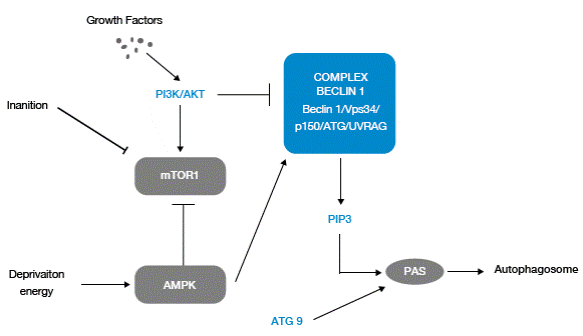
Figure 1 Molecular regulation of autophagy. Source: Own elaboration based on the data obtained in the study.
Autophagy responds to downregulation by stimuli of growth factors that regulate the phosphatidylinositol-3-kinase pathway (PI3K/AKT), which controls the activation of the mTOR pathway; the latter resides in a multi-protein and macromolecular (mTORC1) complex that is activated by signals associated with nutrients, including amino acids and growth factors, and downregulates autophagy by interacting with the complex Beclin 1.
Autophagy also responds to control due to cellular energy depletion through increased activity of protein kinase activated by AMP (AMPK). In response to elevated levels of 5'-monophosphate adenosine AMP, the inactive AMPK mTORC1 and active Beclin 1 promote Atg9 traffic.
Beclin 1 is associated with a macromolecular complex, which includes hVps34, PI3KC3 class III, p150 and UVRAG. The Beclin 1 complex produces phosphatidylinositol-3-phosphate (PI3P), which recruits factors associated with autophagosome formation.
For inhibition of autophagy, the serine/threonine kinase mTOR protein is the most important in human cells; inhibition occurs through maintenance of hyper-phosphorylation of proteins that are needed for initiating the autophagy signaling pathway. mTORC1 promotes protein synthesis, cell division and metabolism in response to the availability of nutrients, growth factors and hormones, while suppressing autophagy. Mutations acquired in different regions of the mTOR C-terminal promote its hyperactivation, benefiting the uncontrolled growth of tumor cells 2.
Types of autophagy
There are three main types of autophagy that work for eukaryotic cells: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA), which are all different in terms of the mechanics of the process (Table 1).
Autophagy and its mechanism as a tumor suppressor
Although autophagy is a survival pathway -used by both normal and tumor cells to survive hunger and stress- paradoxically, its defects are found in many types of human tumors. Allelic loss of gene Beclin 1, essential for autophagy, is common in human breast, ovarian and prostate cancer 16.
In early stages of the tumor, autophagy acts as a tumor suppressor process, since it is responsible for inhibiting the inflammatory events associated with cancer, and it also promotes genomic stability 17. Furthermore, the accumulation of reactive oxygen species (ROS) is one of the main consequences of metabolic stress, which can cause damage to the structure of DNA through the induction of double strand breaks and change in the base sequence of the DNA, leading to activation of proto-oncogenes and, simultaneously, inactivation of tumor suppressor genes 18.
Table 1 Types of autophagy
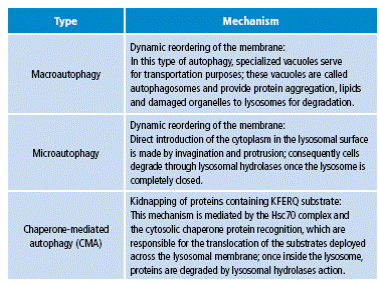
Source: Own elaboration based on Yorimitsu & Klionsky 14 y Chen & Klionsky 15.
For several years, ROS have been linked to cancer development in humans; after many studies, it has been concluded that autophagy plays an important role in reducing levels of ROS 19. The existence of high levels of ROS activates autophagy to eliminate these harmful compounds and, in consequence, prevent DNA damage and the development of tumorigenesis 18.
The study by Cao B et al. 20 shows that anti-microbial agents such as clioquinol, can induce tumor cell death; basically, its anti-tumor properties are given by their ability to activate autophagy in cancer cells by increasing the PI3KC3/Beclin 1 complex and disrupting the mTOR signaling pathway.
Autophagy and its mechanism as tumor inducer
One of the most remarkable abilities in the repertoire of tumor cells is the activation of autophagy in response to stress, which allows long-term survival, particularly when apoptosis is defective. Apoptosis might normally eliminate stress resistant tumor cells as a tumor suppressor mechanism; however, tumor cells often evolve to generate defects in this process, allowing activation of autophagy to sustain the survival under nutrient deprivation conditions. Tumor cells can digest themselves gradually under prolonged stress, becoming less than a third of normal size (Figure 2) 21.
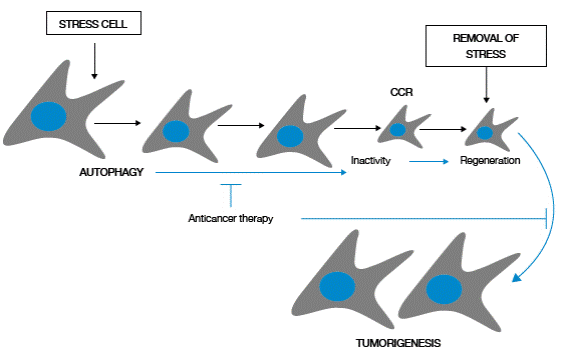
Figure 2 Survival and regeneration mediated by autophagy in tumor cells. Source: Own elaboration based on the data obtained in the study.
Cellular stress activates autophagy in tumor cells, allowing survival by promoting selection of material for cell consumption. As a consequence, small cells that may remain in a dormant state in the presence of stress are generated, but these cells, capable to recover (complete cytogenetic response-CCR) regenerate and restore their cellular proliferation when stress disappears.
Establishing cell latency as a regenerative capacity is highly dependent on autophagy; in tumor cells with defects in autophagy, achieving latency and cell regeneration is less efficient. Therefore, autophagy confers tumor cells tolerance to cellular stress, limiting damage and maintaining cell viability (Figure 3) 22.
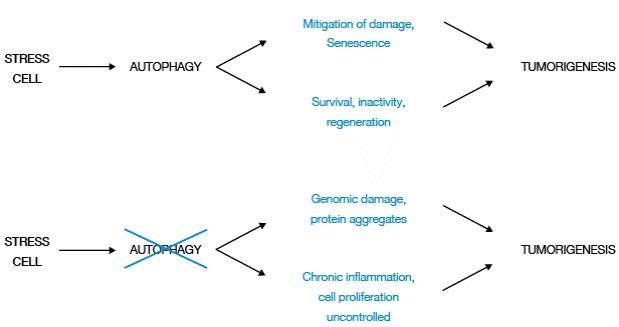
Figure 3 Two-way function of autophagy in tumorigenesis. Source: Own elaboration based on the data obtained in the study.
Although autophagy mitigates damage and promotes cellular senescence, inhibiting tumorigenesis by allowing tumor cells to survive cellular stress, remain dormant and regenerate with elimination of stress, it also promotes tumorigenesis.
Other factors that can originate and stimulate tumorigenesis are damaged tumor cells in tumors with defects in autophagy, particularly those with protein aggregates and genomic damage, and the presence of chronic inflammation, which generates a favorable microenvironment that alters cell death.
Targeting autophagy for cancer therapy
The role of autophagy in oncogenesis is variable, since it is a tumor suppressor during the early stages of the tumor and may contribute to their growth during their development 23. Autophagy, as a response to cancer therapy, can promote/suppress tumor development, for this reason, improving cancer therapy is considered an unusual goal.
Tumor cells with competent autophagy
This type of cells can activate autophagy as an adaptive response to therapeutic agents against cancer, therefore, autophagy could act as a resistance or survival mechanism of tumor cells in prolonged treatments. The absence of cancer cells in an essential mechanism for resistance, by specifically inhibiting autophagy, is expected to improve the efficacy of anticancer drugs 24. Cancer cells with defective apoptosis and low metabolic stress have been proved to establish autophagy activation as a survival mechanism; in contrast, tumor cells with competent apoptosis under stress may undergo rapid cell death following activation of apoptosis. Therefore, inhibition of autophagy is expected to be therapeutically more beneficial in the treatment of tumors that have defects in apoptosis 25.
Autophagy inhibition: therapeutic target against cancer
Inhibition of autophagy as a scenario to sensitize tumor cells to anti-neoplastic treatment has been validated in several studies. Inhibition of autophagy by chloroquine, a lysosomotropic agent which raises pH and interferes with autophagosome degradation within lysosomes, shows an improvement in anti-tumor activity of cyclophosphamide (alkylating agent) in a lymphoma induced model and colorectal cancer 26. Similarly, 3-methyladenine (3-MA), an autophagy inhibitor, allows sensitization in nasopharyngeal carcinoma cells (Hone-1) to treatment with cisplatin and radiotherapy, which relates to the prevention of endoplasmic reticulum stress induced by autophagy in such cells 27.
Several studies support the idea that autophagy, as a physiological process in response to treatments in cancer cells, can help tumors evade drug - induced cytotoxicity -as survival mechanism-.Thus, it was demonstrated in non-small cell lung cancer (NSCLC) that autophagy regulates their resistance to treatment with paclitaxel, mainly by decreasing microRNA-216b (miR-216b); therefore, strategies that increase levels of miR-216b or inhibit cell autophagy can improve the outcome of treatment with paclitaxel against NSCLC 28. Furthermore, it has been shown that chemoresistance of patients with hepatocellular carcinoma (HCC), compared to treatment with cisplatin, is the consequence of activation of autophagy by binding lectin beta-galactoside, and galectin-1 29.
In order to determine the molecular mechanisms of the chemotherapeutic effect of chloroquine on malignant glioblastomas, recent studies have been devoted to probing the cytotoxicity of chloroquine in combination with temozolomide (TMZ), taking ROS as one of the main causes of dysfunctional mitochondria.
Hori YS et al. found that chloroquine increases cellular ROS and cytotoxicity of TMZ in glioma cells by inhibiting mitochondrial autophagy 30,31. Also, recent studies in patients with positive estrogen receptor (ER (+)) showed a significant increase in sensitivity to apoptosis in breast cancer induced by tamoxifen and fulvestrant after inhibition of autophagy induced by microRNA 214 (miR-214). These results support the regulation of autophagy as a new therapeutic strategy for overcoming endocrine resistance in breast cancers ER (+) 32.
Autophagy is commonly regulated in tumor and normal cells exposed to cancer therapies, but the greater dependence of tumor cells -compared with normal cells- on the cytoprotective effects of autophagy offers a new therapeutic opportunity. In fact, autophagy is induced as a strategy for survival in human tumor cells treated with histone deacetylase inhibitors (HDAC) 33, arsenic trioxide 34, tumor necrosis factor alpha (TNF α) 35, gamma interferon (IFN-γ) 36, rapamycin 37 and hormone therapy antiestrogen 38, suggesting that inhibition of autophagy could reduce resistance of cancer cells in these therapies.
Another strategy for inhibition of autophagy includes the use of siRNAs (small interfering RNA), which target autophagy essential genes and sensitize cancer cells to the induction of cell death by radiation cells 39, and a wide range of chemotherapeutic agents, including cyclophosphamide and N-(4-hydroxyphenyl) retinamide 40.
Tumor cells with defective autophagy
These tumors probably adapt to a state of defective autophagy over time and acquire compensatory mechanisms of cell survival. Hence, cancer cells with defects in autophagy are not expected to rely on this mechanism for cytoprotection during chemotherapy and radiotherapy; inhibition of autophagy cannot increase cytotoxicity by anti-neoplastic or irradiation drugs 41.
Moreover, tumor cells with defective autophagy probably have high susceptibility to metabolic stress, high levels of DNA damage and propensity to genomic instability, which are properties with different implications for responsiveness to anti-neoplastic treatments 42-43. Although, it is still poorly documented, tumor cells with defective autophagy may be particularly sensitive to metabolic stress induction regimens, such as anti-angiogenic pharmaceutical drugs, growth factors receptor inhibitors, glucose deprivation and agents that induce DNA damage, including platinum compounds and topoisomerase inhibitors 44.
Induction of autophagy: therapeutic target against cancer
Since defects in apoptosis are often observed in many tumor cells and may increase their resistance to several conventional therapies for carcinogenesis, targeting alternative pathways to cell death is an attractive strategy to improve anti-tumor therapy 45. Consequently, induction of autophagic cell death can serve as a novel therapeutic strategy to eliminate the development of various cancers, especially those with high thresholds of deficient apoptosis 46.
Several studies have reported that different agents, including arsenic trioxide 46 and the vitamin D analog EB1089 47, induce autophagic cell death in tumor cells in vitro; unfortunately, in these cases, autophagic cell death was determined based on morphological characteristics, so the studies may not represent a true autophagic cell death 48,49. However, other reports have shown specific examples of autophagic cell death in response to certain agents.
Some tumor cells, especially those lacking essential modulators of apoptosis such as BAX, BAK or caspases, exhibit cell death autophagy in vitro when treated with certain chemotherapy drugs, such as etoposide, fenretinide and dexamethasone 50-52.
Also, other studies have shown that polyphenols activate autophagy, controlling cell regulator mechanisms; these results provide strong support to the idea that plant polyphenols are really useful in treating diseases such as cancer, where autophagy plays an important role 53.
Induction of autophagy, in response to nutrient starvation, is responsible for the beneficial effects on longevity in the presence of caloric restriction; at least in Caenorhabditis elegans, its activation slows aging and prolongs useful life 54.
It is tempting to speculate that periodic induction of autophagy may also be responsible for a preventive effect against the development of cancer processes in the presence of caloric restriction; if this were true, the pharmacological induction of autophagy could be used for chemoprevention of cancer. Future studies are needed to clarify whether the induction of autophagic cell death in cancer has a relevant clinical utility.
Conclusions
Autophagy can act in two ways during cancer development: as a mechanism of tumor suppression or as an adaptive response to stress to maintain cell survival. Nonetheless, the molecular mechanisms underlying the regulation of autophagy and the role of this process in tumor cells is not fully understood yet. For this reason, pharmacological modulation of autophagy may have significant clinical potential as a new therapeutic strategy for the eradication of cancer.
Induction of autophagy may be useful for cancer chemoprevention in normal cells or for triggering an alternative cell death mechanism in certain tumor cells, especially those with compromised apoptotic functions. Furthermore, deletion of autophagy pathway scan be combined with conventional antitumor regimens to achieve greater efficiency, thereby, avoiding drug resistance in tumor cells, which represents a valuable therapeutic strategy for radio and chemo-sensitization.
On the other hand, additional questions and concerns arise, as it is known that autophagy inhibits oxidative stress, inflammation and genome instability, favoring tumor suppression in some models; it is still to determine whether these events contribute to the suppression of human cancer. If so, the essential autophagy gene should be represented among genes with recurrent mutations in the development of human cancers. Based on currently available data, this does not seem to be the case, but there is a possibility that the loss of tumor suppression by autophagy in cancer occurs indirectly.
The use of autophagy against cancer offers new opportunities for drug development, as more potent and specific inhibitors of this process are clearly needed to ensure the efficacy and safety of anti-tumor treatment. Future efforts should focus on the modulation of autophagy for maximum therapeutic benefit, as well as in elucidating the genetic and physiological conditions that determine the function of pro-survival or pro-death autophagy.
A major limitation of the research to date is that all models of cancer have addressed the role of autophagy only in tumors, leaving aside the direct comparison with this deficiency in normal tissues.
Since, there is evidence that autophagy is important for some normal tissues, a critical question is whether the systemic inactivation of this process is selective enough to harm cancer growth without affecting normal tissues with harmful consequences.
Ultimately, the pharmacological manipulation of autophagy for prevention and treatment of cancer depends on the ability to correctly recognize the functional status of autophagy in tumors and the availability of specific modulators.

