











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
With an incidence of 10.6 per 100 000 inhabitants and a mortality rate of 6 per 100 000 inhabitants, colorectal cancer (CRC) is currently the first or second leading cause of cancer-related deaths in Colombia. However, these statistics have increased; in 1997, CRC was considered as the fourth leading cause of cancer deaths and an estimated of about 75 people died from the disease. CRC is the third leading cause of cancer in men and the second in women, with a standardized incidence ratio (SIR) of 83 and 48, respectively, while in developing countries it is the sixth leading cause.
Lynch syndrome is the most common cause of inherited CRC and represents 5-8% of all cases 1-5; it is associated with mutations in the germline of MMR genes including hMLH1, hMSH2, hMSH6, hPMS2, hPMS1 and hMLH3, which generate microsatellite instability in most cases. About 90% of the mutations identified in this group correspond to hMLH1 (50%) and hMSH2 (40%) genes 6.
In a previous study conducted to detect of mutations in MLH1 and MSH2 genes in Colombian families, the detection rate for this disease was 35% 7.
Clinical features
Lynch syndrome is a hereditary cancer syndrome with an autosomal dominant inheritance pattern. 40-60% of families who meet the clinical criteria for this disease have mutations in MMR genes. The risk of developing cancer among mutation carriers is 80% at age 70, and the average age for the onset of a neoplastic lesion, either in the colon or outside the colon, is 45, much earlier than in sporadic cancer; however, the risk of cancer and age of onset is different for each of the genes involved 8,9. Some studies suggest that mutations in MLH1 are at increased risk of CRC in MSH2, which have increased risk of extracolonic cancers. Mutations in MSH6, when compared to MLH1 and MSH2, show a lower expression of CRC, but an excess of endometrial cancer 10.
From the anatomopathological point of view, adenocarcinomas framed in this syndrome are characterized for being solid, poorly differentiated, mucinoide-like, with signet ring cells and peritumoral lymphocytic infiltration -similar to the infiltration observed in Crohn's disease-, which is currently considered as a prognostic marker. The most common site of lesion is the proximal colon and the number of adenomas varies slightly with villous growth 8,11.
Extracolonic malignancies that occur in Lynch syndrome include endometrial, stomach, ovary, ureter, renal pelvis, brain, small intestine, hepatobiliary and skin (sebaceous adenoma) cancers. These tumors can be synchronous or metachronous.
Regarding extracolonic tumors, endometrial cancer predominates in Western countries and gastric cancer in Eastern countries 12; less frequently, cases of breast cancer have also been reported 13.
Classification of clinical criteria
In clinical practice, the diagnosis of Lynch syndrome is mainly based on Amsterdam I criteria. Selecting families using these criteria allows a mutation detection rate of about 60% 14 (Table 1).
Table 1 Amsterdam I Criteria.

CRC: colorectal cancer; HNPCC: hereditary nonpolyposis colorectal cancer; MSI: Microsatellite instability.
Source: Own elaboration based on data obtained from Asad Umar 15.
Amsterdam II criteria were proposed later, in 1999, because the first classification did not include extracolonic tumors, which are part of the phenotype of hereditary nonpolyposis colorectal cancer (HNPCC) (Table 2). Today, the revised Bethesda criteria, established by the US National Cancer Institute, are also taken into account, which allows selection of patients by determining the microsatellite instability 15 (Table 3).
Table 2 Amsterdam II criteria.

CRC: colorectal cancer.
Source: Own elaboration based on data obtained from Allen et al. 18.
Table 3 Revised Bethesda criteria (Asad Umar).

CRC: colorectal cancer; HNPCC: hereditary nonpolyposis colorectal cancer;
MSI: Microsatellite instability.
Source: Own elaboration based on data obtained from Asad Umar 15.
Repair systems
Repair systems are crucial for maintaining the integrity of the genome. The mismatch system repair increases fidelity of replication by a factor of 1 000 correcting errors generated during this event. The process begins by recognizing the alteration of DNA and continues with the repair of the defect. The mismatches are caused by errors during replication and recombination, through the generation of small insertions or deletions or physical damage of DNA caused by deamination or cytosine methylation. The best studied system is mutHLS in Escherichia coli; here, mismatch recognition is performed by the MutS protein with ATPase function 16-19.
MMR system in humans
The MMR has evolved to correct errors that are beyond 3'→ 5' exonuclease correction activity of DNA polymerases. The process begins with the recognition of the mismatch caused by the binding of the hMSH2/hMSH6 heterodimer, also known as hMutSa; this complex undergoes a conformational change promoted by ATP which turns it into a clamp, which is displaceable through the DNA strand and, then, recruits the hMLH1 / hPMS2 heterodimer, also known as hMuLa.
This ternary complex can move in any direction along DNA, and when it encounters the broken chain that is subjected to PCNA loading an exonuclease 5 '→ 3' (EXO1), degradation of the thread starts towards the site where the mismatch is located. The chain that is not degraded, is stabilized by replication protein A (RPA) preventing the action of EXO1. When the lesion is removed, the degraded region is again synthetized by DNA polymerase then the ends are joined by DNA ligase action 20,21 (Figure 1).
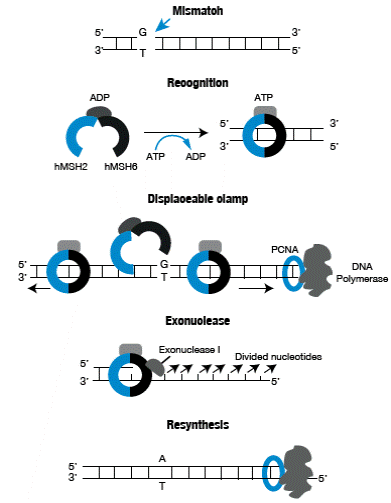
Figure 1 Mismatch repair process. Source: Own elaboration based on data obtained from Boland et al.25.
MLH1 gene is located in the chromosomal region 3p21.3, with a length of 2752pb and 19 exons, and encodes a protein of 756 amino acids with a conserved region of 300 amino acids in its N-terminal end and 27 splicing variants. Location of protein is intranuclear 17,22.
MutL and hMLH1 are members of the GHKL ATPase/kinase superfamily including gyrase, type II topoisomerase, Hsp90 and histidine kinase. The hMLH1 protein has three domains of importance: a ATPase domain at the N-terminal region, an interaction domain with MutS that has another flexible and poorly preserved hinge region, and a domain in the C-terminal region (CTD) involved in homo- and heterodimerization 17. Protein produces heterodimers with hPMS2 proteins, forming the MutLa and hMLH3 complex, and also the MutLb complex which interacts with PCNA.
The hMLH1 protein is part of the surveillance genome complex known as BASC, which includes BRCA1, BLM and ATM proteins, and RAD50-MRE-NBS1, MSH2, MSH6 and MLH1 complexes. They intervene as control points during the cell cycle in the presence of DNA damage 23.
MSH2 gene is located in the region 2p21, is 3307pb long and has 16 exons, encodes a protein of 934 amino acids and has 13 splicing variants. The localization of the protein is intranuclear 22 and contains five domains: domain I DNA binding, domain II interaction with hMSH3 and hMSH6, domain III of ATP binding, domain IV of interaction with homologous MutL, and domain V of ATPase function 24. The protein forms two heterodimers: MutSa, consisting of MSH2/MSH6, which is more involved in the repair process, and MutSb, consisting of MSH2/MSH3.
As mentioned above, the MSH2 protein is also part of the genome surveillance complex and acts as a potential damage sensor in recombination and replication 23.
Microsatellite instability
Microsatellites correspond to short repetitive sequences of 1-4 nucleotides, generally adjacent to coding regions; they are also known as short tandem repeats (STR). Microsatellite instability (MSI) is defined as a change in length of the repeat units due to an insertion/ deletion of one or more of these units. This can occur in a microsatellite tumor tissue when compared to normal tissue from the same patient 25. When there is damage in MMR, microsatellites tend to change the number of repetitions. About 70-90% of cases of HNPCC show positive MSI, which is even higher in families with mutation in any of the MMR genes, whereas, in sporadic colorectal tumors, it is only observed in 10-15% of cases 26; therefore, instability is a relatively sensitive but nonspecific marker for HNPCC 25.
In 1998, the National Cancer Institute of the United States proposed a panel of five markers for MSI analysis: mononucleotide in repeats BAT25 and BAT26 and dinucleotide repeats in D2S123, D5S346 and D17S250. A tumor is graded as high MSI (MSI-H), if two or more markers are altered; mild or low microsatellite instability (MSI-L), if a marker is altered, and stability (MSS) if markers are not altered (Table 4).
Table 4 Criteria interpretation of microsatellite instability.
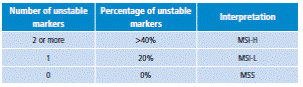
Source: Own elaboration based on data obtained from Boland et al.25.
BAT26 is extremely sensitive for detecting tumors with instability and shows an insignificant size variation, either between two alleles of an individual or between individuals 25.
Colorectal tumors with MSI-H are found predominantly in the proximal colon, have histopathologically mucinous appearance and may be resistant to cytotoxicity induced by chemotherapeutic agents.
Microsatellite instability varies from adenoma to adenocarcinoma and, then, to metastatic tumor.
MSI testing has 80-91% sensitivity in patients with mutations in the MLH1 or MSH2 gene and of 55-77% in MSH6 or PMS2; nevertheless, specificity is similar 27.
MMR gene and somatic mutations in Lynch syndrome
The MMR genes have a role in genome maintenance. The presence of a mutation in some of these genes, mainly MLH1 or MSH2, triggers a cascade of events that affect genes with tandem repeats in their sequence. The repetitive sequences are highly susceptible to misalignment during the replication process, resulting in an increase of 100 times their mutation rate.
Repetitive sequences are dispersed throughout the genome; a large number of human genes have mononucleotide repeats, therefore, they are possible targets of change in the frameshif during replication, which, in turn, generates truncated proteins. These genes, which include MMR, MED-1 and RAD50, are usually involved in signal transduction (TGFβ-RII, IGFIIR, PTEN), apoptosis and inflammation (BAX, caspase-5), transcription regulation (E2F4, TCF-4) and repair 19.
Among the most important genes susceptible to mutations, gene TGFβRII (receptor II transformer of growth factor β) is found, which contains an adenine (A) repetition tract 7 found in 75-90% of patients with MSI, for both HNPCC and sporadic colon cancer. If the inactivation of one of the receptors occurs, the cells lose their responsiveness to TGFβ and cell growth, which represents an important milestone in tumorigenesis of various cancers such as stomach, neck and prostate. In colon cancer, this inactivation corresponds to an event that occurs early during the transition from adenoma to carcinoma. The inactivation of TGFβRII occurs frequently in MSI+ gastric tumors, but is rare in MSI+ endometrial tumors.
Insertion/deletion mutations in repetitive mononucleotide regions are located in BAX (G8), TCF-4 (A9), IGFIIR (G8) and hMSH6 (C8) genes. Such mutations also occur at a significant rate in MSI+ colorectal tumors. Other genes such as caspase-5 (A10), hMSH3
(A8) and RAD50 (A9) are inactivated, with a lower frequency, in primary tumors, but show a high incidence of frameshift mutations in CRC cell lines 19.
In a study by Yamaguchi et al., the frequency of frameshift mutations in genes ACVR2 (activin receptor 2) and TGFβ -RII was between 70-95% for HNPCC. The signaling pathway of TGFβRII downstream includes Smad 2, 3 and 4 proteins, with subsequent inhibition of cell growth; this pathway may be disrupted by mutations in ACVR2. The Wnt pathway is also affected by disruption caused by mutations in APC. Other genes less involved in the early stages of tumor development are PTHLH, MARCKS, hMSH3, TCF4, CASP5, RIZ and RAD50 28.
Molecular analysis for the diagnosis of Lynch syndrome
Studies have been performed worldwide to determine the presence of mutations that predispose to Lynch syndrome, and about 400 genes in all MMR have been identified; 90% are in MLH1 and MSH2. The first publications about Lynch syndrome indicate that age 44 to 45 is the average for CRC installation. However, it is now clear that not all cases appear at such a young age and this can vary according to the type of mutation which may not differ from sporadic cases, and could explain the lack of sensitivity of Amsterdam and Bethesda criteria 29.
The most frequent mutations in MLH1 and MSH2 genes are nonsense, missense, frameshift and splicing site changes, while the proportion of genome rearrangements varies in each population from 5% to 20% on average; there are also low frequency cases (1.5%) and, others with higher frequency due to a founder effect 12,31,32, like the results obtained from a study of Spanish population 30.
Conventional methods have been used for detecting mutations as single stranded conformational polymorphism (SCPP), denaturing gradient gel electrophoresis (DGGE), conformation sensitive gel electrophoresis (CSGE), denaturing high performance liquid chromatography (DHPLC) and direct sequencing of the gene. Since large genome rearrangements are not detected by these methods, the analysis by MLPA (Multiplex Ligation-Dependent Probe Amplification) is used, which allows semi-quantitatively assess the change number of copies in a specific region of the gene. The combination of techniques allows a better characterization of the mutational spectrum in a population study 12,29,33-36.
Given the high cost of molecular screening for all MMR genes in suspected families for Lynch syndrome, several pretest strategies, such as determining the degree of instability in tumors, have been suggested. The MSI phenotype is a specific marker useful in HNPCC. However, this can also be observed in sporadic cancers, since about 15% of them are caused by somatic mutations, loss of heterozygosity of MMR genes and promoter methylation of the gene MLH1; this situation is also observed in a study of hypermethylation of MLH1 promoter, predominantly in women over 40 with CRC 37.
The promoter methylation status is suggested as a marker to distinguish sporadic tumors from hereditary tumors. The discrimination of these two subtypes may improve detection strategy, therapy and prevention. In order to standardize the MSI tests, a working group from the National Cancer Institute in the United States recommended the use of the Bethesda panel with five markers, two single nucleotide and three dinucleotide 38,39. In this regard, Pedroni et al.40 demonstrate that a two mononucleotide markers panel (BAT 25 and BAT 26) is more efficient in detecting tumors with high MSI in the absence of MMR proteins compared with the Bethesda panel (93% vs. 54%).
Another initial approach method is the demonstration of the absence of MMR protein expression through immunohistochemical staining (IHC). It has been proved that IHC for MMR proteins (MLH1, PMS2, MSH2 y MSH6) provides a faster, cost-effective, sensitive and very specific screening technique for MSI. Some studies compare methods for determining MSI based on PCR and IHC, showing high concordance (97.8%) 41-43.
IHC assessment for MMR protein expression is performed on tissue sections containing both tumor tissue and normal colonic mucosa. Monoclonal antibodies of mice are used in different dilutions for the total length of proteins. Normal tissue and lymphocytes adjacent to the respective tumor are used as positive internal controls and the loss of protein expression is defined as the complete absence of nuclear staining in tumor cells, but maintained in epithelial and normal stromal cells 40.
Stojic et al.20 described a group of patients with symptoms compatible with Lynch syndrome in absence of IHC expression of the MSH2 protein and had no detectable mutations in this gene; individuals had deletion of epithelial cell adhesion molecule gene (EpCAM) located upstream of MSH2. In this study, it was also established that this deletion leads to somatic hypermethylation of MSH2 and loss of protein expression. The silencing of MSH2 gene promoter by deletions of gene EPCAM causes Lynch syndrome in 20-25% of patients with IHC negative for MSH2 and in whom no mutation is found in the germline, which corresponds to 2-3% of all patients with this syndrome 20. Studies have shown that a negative IHC for EpCAM with negative MSH2 indicates deletion of EpCAM with 100% specificity 44,45.
Lynch et al.46 suggest that patients with suspected Lynch syndrome should initially have a test for MSI and then immunohistochemistry tests for MMR proteins, and molecular tests for the negative gene of IHC. In addition, the authors recommend that patients with clinical criteria for Lynch syndrome, even in the absence of germinal mutations for MMR, should be monitored and followed just like molecularly similar patients.
In 2011, a survey conducted by the National Society of Genetic Counselors to evaluate screening programs for Lynch syndrome and barriers for their implementation showed that more than 50% of respondents had been subjected to a screening protocol once the first case of colon or endometrial cancer in the family appeared.
Screening methods in tumor tissue varied in 64.2% when the study initiated with IHC testing, 20.8% with MSI testing and 15% with both tests simultaneously. Also, with the results of the survey, the cost of testing and the lack of medical information were deemed as the most important barriers to screening 29.
Moreover, taking into account that only up to 80% of germline mutations are detected in despite of the Amsterdam and Bethesda clinical criteria, there are increasingly strong trends that support universal screening of all newly diagnosed cases with CRC and endometrial cancer 29,47,49.
The debate still lingers regarding the methods used to start screening for Lynch syndrome, as several authors propose IHC as the first test due to its cost-effectiveness and because the absence of protein expression can be detected in both CRC as endometrial cancer. However, as mentioned above, a percentage of tumors with absence of MLH1 and MSH2 expression may relate to somatic events such as promoter hypermethylation or BRAF V600E mutation. Cost-effectiveness data suggest that the best strategy for these cases is to follow IHC test with BRAF mutation or hypermethylation of MLH1 promoter test 48.
Shi et al.49 suggest that, for patients with high microsatellite instability, somatic BRAF V600E mutation should be considered as a pre-molecular study of MMR genes, because this alteration is much more related to sporadic tumors and, thus, Lynch syndrome can be discarded. Similarly, the use of multiplex ligation-dependent probe amplification (MLPA) to detect large rearrangements corresponding to 20% of the mutations is suggested 49.
Liu et al.50 propose a diagnostic strategy for Lynch syndrome that starts with finding MSI, uses the panel of five mononucleotide markers, and searches for specific high prevalence mutations, comprehensive determination of mutations in the MLH1 and MSH2 by sequencing and techniques for large MLPA rearrangements. All of this should be done before searching in other genes, including MSH6 and PMS2, through the MLPA technique 50.
Conclusions
The molecular diagnosis of Lynch syndrome is essential to locate affected individuals and carriers in families and to provide adequate monitoring and genetic counseling. It is necessary to gather evidence on the cost-effectiveness of making universal screening on CRC or to start the process with the use of clinical guidelines, determining the degree of microsatellite instability and IHC, and with this result, determining the next step for sequencing and search of large rearrangements in the MLH1 and MSH2 genes, and subsequently in MSH6, PMS2 and EpCAM.

