











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Autoimmune hemolytic anemia (AHA) is a relatively rare disease with an annual incidence of 1 per 75 000 to 80 000 people. The average age of diagnosis is 3.8 years in the pediatric population 1. For decades, autoimmune entities such as hemolytic anemia, thrombocytopenic purpura, neutropenia and insulin receptor antibodies have been reported as paraneoplastic manifestations of Hodgkin's disease (HD) 1-4. Therefore, all patients with AHA should be studied to discover the cause and to discard the presence of neoplastic based entities such as HD and non-neoplastic entities such as infections, autoimmune diseases, drug intake, etc. 5.
The development of autoimmune hemolytic anemia in lymphoproliferative disorders is multifactorial and caused by disorders in regulatory and autoreactive LB and LT 6 cells. The Hodgkin lymphoma implies alterations in the immune system, which include abnormalities in the cytokine production and increased sensitivity to regulatory T cells, but with an overall decrease in the number and functional capacity of T cells 6-8. Thus, during a decrease of cytotoxic T lymphocytes, an increase in autoantibody production may occur; this is currently the most accepted mechanism to explain the appearance of AHA in some of these patients 9.
In the past, based on epidemiological studies, the hypothesis of a possible infectious etiology during HD that instigates the production of antibodies that cross-react with antigens in the erythrocyte membrane 10,11 was proposed, however, this theory has not been proven yet.
This paper describes the case of a pediatric patient who developed hemolytic anemia as an initial manifestation of Hodgkin lymphoma; this finding is highlighted as a paraneoplastic manifestation of HD that should be taken into account when studying the causes of AHA.
Case presentation
Male patient, 11 years old, with a history of anemia of 10 months which was diagnosed in a control consultation, with unclear characteristics and etiology, and managed with ferrous sulfate.
On admission in hospital, the relatives referred fever peaks of up to 39°C, predominantly nocturnal, associated with asthenia, adynamia and unquantified weight loss. Physical examination showed a thin aspect and muco-cutaneous pallor. A complete blood count found hemoglobin at 7.4 g/dL, mean corpuscular volume of 77.7fl and mean corpuscular hemoglobin of 25.9pg; the blood smear showed moderate anisocytosis with presence of dacrocytes and codocytes. The patient was hospitalized under the diagnosis of normocytic normochromic anemia of unclear etiology; studies were extended and a direct Coombs test, with positive results (+++), and a high LDH were performed, which finale led to conclude that the patient presented immune hemolytic anemia.
The fever and weight loss manifestations suggested a neoplasm as the cause of the immune hemolytic anemia; therefore, further studies, such as chest radiography and abdominal ultrasound, were performed. The first exam showed no alterations and the second showed para-aortic lymphadenopathy and left iliac chain, as well as hypo-echoic images in the common left iliac artery. A biopsy and bone marrow aspiration were also performed, which reported no tumor infiltration. Due to the presence of an acute hemolytic picture, treatment with methylprednisolone 30 mg/kg/ day for three days was ordered; after this, a control complete blood count was performed, in which an increase in the value of Hb 8.9g/dL was found. Treatment was continued with prednisone at 1 mg/kg/day.
The studies initially performed to find a possible infectious, tumor and autoimmune disease causes were negative. The patient persisted with spiking fevers and presented painful hepatomegaly during the clinical evolution, so a new abdominal ultrasound was requested, in which enlarged lymph nodes in the para-aortic region with involvement of the left iliac region were found. The child was further evaluated through pediatric surgery for lymph node biopsy; the pediatric surgeon ordered complementary studies including an abdominal computed tomography scan that reported lymphadenopathies in the left iliac chain with ipsilateral para-aortic region involvement. Based on these findings, a magnetic resonance imaging of the abdomen and pelvis was ordered, which showed splenomegaly, nodes in the para-aortic region, left infrarenal aortoiliac bifurcation and solid smooth masses accompanying the major vessels and the lower region of the renal hilum, compatible with neoplasia.
A biopsy of the retroperitoneal lymph node showed infiltration of Reed Stemberg cells with positive reactivity in immunohistochemistry for CD15, CD30, PAX-5 and LMP-1 studies, which confirmed the diagnosis of a mixed cellularity Hodgkin disease. The clinical status corresponded to stage IIB, for which chemotherapy with ABVD scheme was started.
The patient received six cycles of ABVD chemotherapy (doxorubicin, bleomycin, vinblastine and dacarbazine) and subsequent consolidation treatment with radiotherapy in sites involved at diagnosis.
At the end of the study, the child was in clinical remission for two years, with control blood counts within normal limits, and monitoring and control through the oncohematology service.
Discussion
Previously, prevalence of autoimmune hemolytic anemia has been reported in patients with Hodgkin's disease ranging between 0.2% and 2.7% 2,12; with the development of more sensitive immunoassays, it is likely that a larger number of HD patients are being diagnosed with AHA 13. However, this type of anemia as the first manifestation of this disease is even more unusual than their mere association 10,14.
Bowdler & Glick 15 first published about this relationship, stressing that the diagnosis of AHA preceded by three years the dignosis of HD, as in the case of this patient, who had anemia for 10 months before diagnosis of Hodgkin lymphoma.
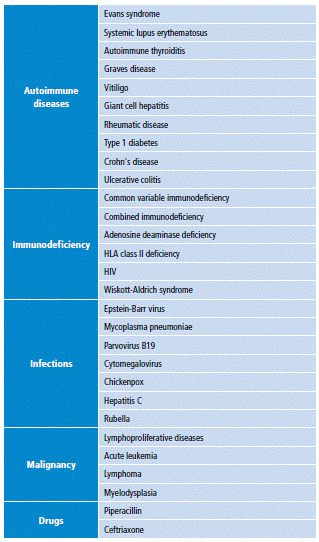
The secondary AHA, besides presenting the characteristic clinic of hemolytic anemia such as dyspnea, fatigue, paleness or jaundice, among others, are presented along with the underlying disease clinic. In the case of a lymphoproliferative syndrome, associated manifestations usually include appetite and weight loss, night sweats, lymphadenopathy and hepatosplenomegaly; this clinic is important because it directs or allows suspecting a neoplastic entity 6,14,16, as in this patient. Moreover, even in the absence of clinical manifestations suggestive of a lymphoproliferative syndrome as the cause of autoimmune hemolytic anemia, this type of pathologies should always be sought once the diagnosis is made, as well as other possible causes (Table 1) 16,17.
Autoimmune hemolytic anemia may be present at any stage of Hodgkin's disease; although it is usually associated with the active or advanced disease, it can precede the diagnosis or be present in a relapse episode 1,10. A positive Coombs test can be an indicator to suspect possible relapses in patients with a history of HD in remission with or without AHA 6,10.
Currently, there are no studies of sufficient quality to establish appropriate protocols for the management of AHA, nor a consensus on what complete remission or partial remission of the disease means 19, therefore, the management of hemolytic autoimmune anemia is based on experience and individual clinical decisions.
It should be noted that the effectiveness of therapies for AHA is low when it is secondary to an underlying disease, this being especially true for autoimmune hemolytic anemias secondary to lymphoproliferative syndromes 6,13.
The above statement was evident in the case reported here, in which no adequate response to treatment with corticosteroids was obtained. Definitive therapy for autoimmune hemolytic anemia associated with Hodgkin's disease is the treatment of the underlying disease, through which there is a progressive decrease of antibody titers until reaching the eventual negativization of the Coombs test 20, the recovery of Hb values and the disappearance of hemolysis signs.
Conclusions
Autoimmune hemolytic anemia, although unusual, can be a paraneoplastic manifestation of a lymphoproliferative syndrome as Hodgkin's disease. The underlying cause of AHA must always be sought, even if it is refractory to treatment. Additionally, if there is an AHA in a patient with a history of HD in remission, HD should be suspected and a relapse should be discarded.

