











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hemofilia A es una enfermedad hereditaria recesiva ligada al cromosoma X que se presenta por la deficiencia cuantitativa del factor VIII de la coagulación. Esta enfermedad afecta a 1 de cada 5 000 a 10 000 varones y se ha reportado que esta incidencia no varía entre poblaciones 1. La enfermedad se caracteriza por la amplia heterogeneidad de alteraciones en el gen del factor VIII de la coagulación 2, el cual se encuentra en la banda más distal del brazo largo en el cromosoma X en la región Xq28, tiene una extensión de 186kb y comprende 26 exones. De estos, 24 varían en longitud de 69 a 262 pares de bases (pb) y los 2 restantes correspondientes al exón 14 y 26 tienen una longitud de 3 106pb y 1 958pb, respectivamente. Además, en la región 3', el exón 26 cuenta con secuencias que no son traducidas.
En pacientes con hemofilia A severa, la mutación más frecuente es la inversión del intrón 22. En este intrón se encuentra la región int22h1, que presenta elevada homología con dos regiones distales al gen del factor VIII: int22h2 e int22h3, localizadas cerca de 500kb de la región telomérica del brazo largo del cromosoma X. Esta inversión patológica resulta de una recombinación homóloga entre las regiones antes descritas, lo que genera la inversión de los exones 1 al 22 en una región externa del locus del factor VIII y, por lo tanto, la pérdida total en la expresión y función de la proteína. A su vez, esto produce un fenotipo severo de la enfermedad. Respecto a la inversión del intrón 1, la región int1h2 se encuentra cerca de 140kb del telómero y se diferencian solo en un nucleótido de la secuencia int1h1 del intrón 1, ubicado en orientación contraria a este. La recombinación intracromosómica de estas regiones resulta en el desplazamiento del exón 1 por 140kb en dirección al telómero. Esta inversión es responsable del 1% al 5% de los casos severos.
La severidad de la enfermedad se clasifica de acuerdo con la cantidad de factor VIII residual, determinada por la actividad enzimática y categorizada en los siguientes intervalos: <1% de actividad se define como severo, entre 1-5% de actividad, moderado, y entre 5-40% de actividad, leve 1. En pacientes con hemofilia A severa, la mutación más frecuente es la inversión del intrón 22 (INV22) 3, seguida por deleciones de grandes segmentos del gen e inversión del intrón 1. Una de las complicaciones más graves de la hemofilia severa es el desarrollo de anticuerpos inhibidores, cuya incidencia estimada en paciente con hemofilia severa está entre 10% y 50% de los pacientes 3-6. En el informe de la Cuenta de Alto Costo y el Ministerio de Salud de 2015 7, se informaron en Colombia 1 325 casos de hemofilia estudiados para inhibidores, 335 de ellos con presencia de anticuerpos inhibidores contra factor VIII.
Según el protocolo de diagnóstico molecular de la enfermedad recomendado para los diferentes centros especializados a nivel mundial, el primer paso en el estudio de mutaciones asociadas es la detección de la INV22 por la frecuencia de presentación, que es cerca del 50% de los casos con fenotipo severo 3,8. Una vez realizado este análisis, si es negativo, se recomienda el estudio de la inversión del intrón 1 (INV1) responsable del 1-5% de los casos severos. Para los casos negativos, se aconseja ejecutar un tamizaje general de posibles modificaciones en otros exones, en los sitios de reconocimiento de corte: splicing y el análisis de la región del promotor 9.
En el presente estudio, se determinó la frecuencia de la INV22 e INV1 del gen del factor VIII de la coagulación en un grupo de pacientes pediátricos con hemofilia A severa y moderadamente severa (actividad del FVIII 1%-2%), con el uso de técnicas moleculares de reacción en cadena de polimerasa de larga distancia (LD-PCR). Del mismo modo, se buscó correlacionar la detección de la INV1 e INV22 del gen del factor VIII de la coagulación con las características fenotípicas de la enfermedad y la presencia de anticuerpos inhibidores en la población de estudio.
Materiales y métodos
Se incluyeron 30 varones menores de 18 años con diagnóstico de hemofilia A de fenotipo severo: pacientes con hemofilia A severa (actividad del FVIII <1%) y moderadamente severa (entre 1% y <2% de actividad de FVIII, medida por método coagulométrico de dos pasos), a quienes se les realizó la prueba para genotipo de las inversiones de los intrones 22 y 1. La información clínica se recolectó de forma retrospectiva. Se midieron variables sociodemográficas, clínicas y de ayudas diagnósticas. A partir de los instrumentos de recolección, se construyó una base de datos en el programa Excel, donde se digitó la información. Para el análisis de las variables, se tuvo en cuenta su distribución. Las variables cuantitativas se presentaron en medidas de tendencia central y de dispersión y las cualitativas, en frecuencias absolutas y relativas. Para esto se utilizó el programa estadístico Stata 11.
Toma y procesamiento de la muestra
Se efectuó una toma y recolección de muestras propias del seguimiento clínico de los pacientes. Una vez recolectada la muestra, se procedió a realizar aislamiento de leucocitos con la metodología de separación por gradientes de sucrosa usando el reactivo LSM (Lymphocyte Separation Medium). El pellet de células obtenido se almacenó a -80°C hasta su procesamiento.
Extracción de ADN
El ADN se extrajo a partir de los pellets almacenados a -80°C y se utilizó el kit PureLinkTM Genomic DNA (Invitrogen), de acuerdo con las instrucciones del fabricante. La pureza y calidad de cada una de las extracciones se estableció a través de espectrofotometría, con una lectura de 260nm y mediante la relación 260/280nm, siendo aceptables relaciones por encima de 1.5. Esto se midió con ayuda del equipo Thermo Scientific NanoDrop 2000c.
Detección de la inversión del intrón 1 y 22 en el gen del FVIII
Se implementó el método basado en LD-PCR (10,11), una de las técnicas para la detección de estas inversiones, entre otras, como la Inverse Shifting PCR 12.
Análisis molecular
La INV22 se genera a través de recombinación homóloga entre int22h1 (región intragénica) con int22h2 e int22h3 (regiones extragénicas) 13. Para estudiar la inversión del intrón 22, se sintetizaron los primers P, Q y A, B, según la metodología propuesta por Liu et al.11. Los primers P y Q se unen a la región int22h1, mientras los A y B se unen a int22h2 e int22h3. Las secuencias usadas en la detección de la INV22 se muestran en la Tabla 1.
Tabla 1 Detalle de las secuencias de los primers usados en la detección de la inversión 22 del gen del FVIII de la coagulación.

Nota: En la publicación original 11, la secuencia del primer B difiere de la secuencia de referencia. Por esta razón, se hace una corrección en la última base.
Fuente: Elaboración con base en 11.
Para detectar la INV22 con LD-PCR, se modificó el protocolo original 11. Debido a la complejidad de la reacción, los mejores resultados se obtuvieron mediante la reacción con mezclas de parejas de primers, de acuerdo con lo reportado por Polakova et al.13. En la mezcla de PCR se usó el kit Invitrogen Platinum® Taq DNA Polymerase High Fidelity, según el cual cada reacción debía contar con los siguientes componentes: Buffer High Fidelity PCR 10X concentración en reacción 1X y mezcla de dNTP 0.5mM, MgSO4 0.2mM, para las parejas de primers P/Q 0.4uM y A/B 0.2uM. En el caso de las parejas P+B y A+Q 0.4uM, se usó 7% de DMSO, 200ng de DNA template y enzima Platinum® Taq DNA Polymerase High Fidelity 1U por reacción.
En esta investigación, se modificó el protocolo propuesto por Liu et al.11 y por Polakova et al.13, por lo que se suprimió 7-deazaGTP. Esto, ya que, a partir de la estandarización, se estableció que este componente no aportaba precisión o mejores resultados en las condiciones de PCR antes mencionadas. Las condiciones de temperatura para la PCR se dispusieron de acuerdo a las recomendaciones del fabricante de la enzima usada. De esta manera, se realizó una denaturación inicial de 94°C por 2min, seguido de 35 ciclos de denaturación a 94°C por 30s, alineamiento a 55°C por 30s y una extensión a 68°C por 13min en cada ciclo. Estas reacciones se llevaron a cabo en el termociclador c1000 Touch Thermal Cycler (BIO-RAD). Como control interno de la reacción en la detección de la INV22 del gen del FVIII, se corrió la reacción con la pareja de primers A/B. Esta se amplificó con y sin inversión, ya que, aunque se generó la inversión, por lo menos una copia de estas regiones externas permaneció intacta (int22h2 o int22h3).
A fin de detectar la INV1, se llevó a cabo el protocolo descrito por Bagnall et al.10. Se usó la mezcla de primers 9cR, 9F e int1h2F en la detección de la región int1h1 y la mezcla int1h2F, int1h2R y 9F para la detección de la región int1h2. Las secuencias que encontraron la INV1 se muestran en la Tabla 2. La expresión del primer exón en pacientes con el gen del FVIII de la coagulación con INV1 no es esperado. A fin de detectar la INV1, se compararon los productos de amplificación de int1h1 e int1h2.
Tabla 2 Detalle de la secuencia de los primers usados en la detección de la inversión 1 del gen del FVIII de la coagulación.

Fuente: Elaboración con base en 10.
Del mismo modo, se empleó el kit Invitrogen Platinum® Taq DNA Polymerase High Fidelity para mezclar la reacción. Según este, cada reacción debe contar con los siguientes componentes: Buffer High Fidelity PCR 10X concentración en reacción 1X, mezcla de dNTP 0.2mM, mezcla de primers en cada caso a una concentración de 0.4uM, DMSO 5%, 200ng de DNA template y enzima Platinum® Taq DNA Polymerase High Fidelity 1U por reacción. Se realizaron dos reacciones por paciente con las mezclas de primers antes mencionadas. Las condiciones de temperatura de la PCR se dispusieron según las especificaciones del fabricante de la enzima: se usó una denaturación inicial a 94°C por 30s, seguida de 30 ciclos compuestos de una denaturación a 94°C por 30s, alineamiento a 55°C por 30s y una extensión a 68°C por 2min. Estas reacciones se llevaron a cabo en el termociclador C1000 Touch Thermal Cycler BIO-RAD.
Luego, el análisis de los productos se realizó en un gel de agarosa al 1%, con uso de buffer de carga BlueJuiceTM Gel Loading Buffer (Invitrogen) y marcador de peso molecular 1Kb Plus DNA Ladder (Invitrogen) y se leyó en el fotodocumentador de imágenes Syngene®, software GeneSnap®.
Se validó la técnica para la detección de la INV1 e INV22 del gen del FVIII tres veces, con controles positivos (varones afectados), proporcionados por los investigadores De Brasi y Lillicrap y controles negativos (individuos sin alteraciones en la coagulación) y solo para efectos de la validación de la técnica mujeres portadoras (heterocigotas para cada mutación).
El protocolo fue sometido a evaluación y aprobación por comités de ética y de investigación de cada una de las instituciones que participaron en el estudio, posterior a la aprobación del comité de ética de la Facultad de Medicina de la Universidad Nacional de Colombia, sede Bogotá. Todos los participantes del estudio diligenciaron el formato de consentimiento informado para cada institución de acuerdo con los principios de la declaración de Helsinki 14.
Resultados
Características demográficas de la población de estudio
En el estudio se analizaron 30 pacientes con diagnóstico clínico de hemofilia A de fenotipo severo, con un rango de edad comprendido entre 0.8 meses a 18 años y una mediana de 11 años. La mediana de edad de diagnóstico de la población de estudio fue de 11 meses con rango de 3 a 120 meses. Las características demográficas se describen en la Tabla 3.
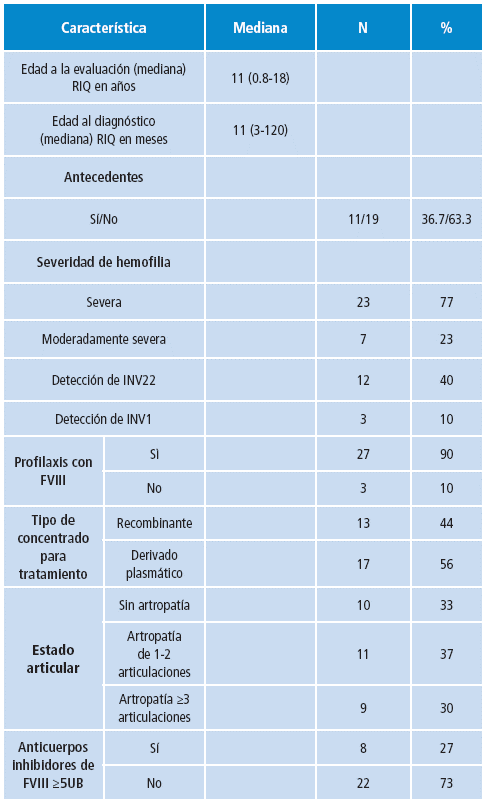
Análisis molecular de la inversión del intrón 22 e intron 1 del gen del FVIII de la coagulación
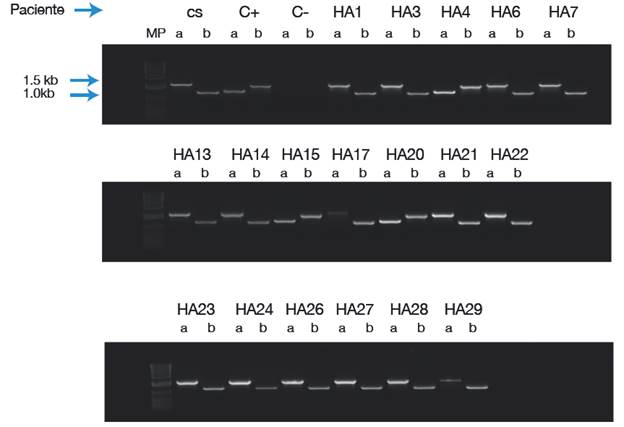
La INV22 se detectó en 12 pacientes (Figura 1) y la INV1, en 3 (Figura 2).

Fuente: Elaboración propia.
Figura 1 LD-PCR para la inversión 22 del gen del FVIII de la coagulación en la población de estudio a partir de la mezcla de primers P+ B y A+Q, los cuales generan un producto quimérico que permite la detección de la inversión. CS: control sano; C+: control positivo; C-: control de reactivos; MP: peso molecular (con el marcador de peso molecular 1Kb Plus. DNA Ladder Invitrogen). Casos positivos para la INV22: HA 2, 5, 8, 9, 10, 11, 12, 18, 19, 25, 30, 31.
Fuente: Elaboración propia.
Figura 2 Detección de la INV1 en la población de estudio. CS: control sano; C+: control positivo; C-: control de reactivos; MP: peso molecular (con el marcador de peso molecular 1Kb Plus. DNA Ladder Invitrogen). Casos positivos para INV1: HA 4, 15 y 20.
Se analizó el desarrollo de anticuerpos inhibidores. El 26.6% de la población analizada (n=8) había desarrollado anticuerpos inhibidores de alta respuesta a la fecha del análisis (Tabla 4). Todos tenían hemofilia A severa. De los 8 pacientes en quienes se detectaron anticuerpos inhibidores a la fecha, 5 (62.5%) fueron positivos para la INV22, 1 (12.5%), para la INV1 y en 2 (25%) no se encontraron las mutaciones en estudio.
Tabla 4 Características clínicas de seguimiento de los pacientes con INV22 e INV1.

Fuente: Elaboración propia.
En el análisis de la relación entre el desarrollo de anticuerpos inhibidores y los pacientes positivos para las inversiones de los intrones 1 y 22 del gen del FVIII, 5/12 pacientes con la INV22 (41.6%) desarrollaron anticuerpos inhibidores y de los pacientes en quienes se detectó la INV1, 1/3 tuvo inhibidores. En 7/12 pacientes (50%) con INV22 y en 1/3 con INV1hubo antecedentes familiares de hemofilia.
Los valores de actividad del factor VIII tuvieron una mediana de 0.8% (rango entre 0.2% y 0.9%) en los pacientes con INV22, se encontró una media de 0.5% (rango entre 0.4% y 0.6%) en aquellos con INV1 y la mediana de actividad fue de 0.8% (rango 0.3% a 2%) para quienes no se detectó mutación.
De los 15 pacientes con INV22 e INV1, 10 recibieron tratamiento con profilaxis con concentrados de FVIII, la gran mayoría, profilaxis secundaria (9/10). De los 15 pacientes sin mutaciones identificadas, 11 recibieron profilaxis secundaria. De los 6 pacientes con inversiones identificadas y anticuerpos inhibidores, se halló que 2 recibieron concentrados de coagulación recombinantes y 4 derivados plasmáticos. De los 13 pacientes que recibieron tratamiento con FVIII recombinante, 2 (15.3%) desarrollaron inhibidores y 6/17 (35.3%) de los pacientes tratados con derivado plasmático tuvieron inhibidores contra el FVIII (Tabla 4).
Respecto al compromiso articular, se encontró artropatía en 20 de los 30 pacientes (66.6%), 14/2 con enfermedad severa, 5/20 con anticuerpos inhibidores. 10/20 pacientes con artropatía exhibieron las mutaciones estudiadas; 8 pacientes, inversión intrón 22, y 2 pacientes, inversión intrón 1.
Discusión
En el presente estudio se analizó por primera vez en Colombia la frecuencia de la INV1 y la INV22 en el gen del FVIII de la coagulación en una cohorte de niños con diagnóstico de hemofilia A, mediante técnicas de LD-PCR. Se logró determinar que estas mutaciones causaron el fenotipo de la enfermedad en la mitad de la población estudiada a través metodologías validadas con anterioridad. El uso de la técnica LD-PCR en la detección de tales mutaciones posibilitó la obtención de resultados reproducibles y fácilmente evaluables, constituyendo una herramienta útil para la detección de la INV1 e INV22 12,13. La INV22 tuvo mayor frecuencia en 12/30 (40%) y la INV1 fue detectada en 3/30 (10%). Estas frecuencias fueron comparadas con diferentes poblaciones, en donde se usó la misma metodología y se observó consistencia entre los datos obtenidos con respecto a la INV22, la cual se ha estimado como la mutación con mayor ocurrencia en pacientes con hemofilia A, equivalente a alrededor de 50% de los casos severos 8,15,16.
Sin embargo, con respecto a la INV1, el porcentaje obtenido en la población del presente estudio (10%) fue superior a la frecuencia observada en otras poblaciones, entre ellas Venezuela y México, con 0% 8,16. Esta diferencia en las frecuencias puede deberse al número pequeño de pacientes evaluados. Uno de los pacientes con enfermedad moderadamente severa tiene INV1, fenotipo de enfermedad severa, con artropatía de dos articulaciones y está en profilaxis. Una posible explicación de la clasificación de severidad de este paciente con actividad de FVIII entre 1% y menos de 2% es la sensibilidad del reactivo que mide la actividad del FVIII, pues en algunos casos no se puede discriminar actividad del FVIII por debajo del 1%.
El 60% de la población estudiada no tiene antecedentes familiares reconocidos de hemofilia, lo que quizá constituye casos de mutaciones de novo. No obstante, es necesario el análisis de la madre y hermanas, según corresponda en cada caso, para comprender los posibles mecanismos genéticos asociados a la aparición de la enfermedad en estas familias.
La principal complicación del tratamiento con concentrados de FVIII recombinante o derivado de plasma, en especial en paciente con hemofilia severa, es el desarrollo de aloanticuerpos inhibidores dirigidos contra FVIII. La prevalencia de estos anticuerpos reportada en la literatura está alrededor de 30%. En un estudio de población argentina 20, se encontraron estos en 18% de pacientes con hemofilia A severa. El desarrollo de anticuerpos inhibidores está asociado a una amplia gama de factores genéticos y no genéticos. Dentro de los factores genéticos, entre los que se destaca el genotipo del FVIII 21 -entendido como la estructura y características del gen dadas las mutaciones presentes hay mayor riesgo de desarrollo de inhibidores en pacientes con grandes deleciones, pues las inversiones de los intrones 22 y 1 son mutaciones de riesgo intermedio para el desarrollo de inhibidores. La historia familiar de persona con hemofilia con anticuerpos inhibidores y la etnia (población no caucásica con mayor frecuencia de inhibidores) son factores relacionados con el desarrollo de anticuerpos inhibidores. Respecto a los factores no genéticos, se hallaron el esquema del tratamiento suministrado en cuanto a intensidad y dosis, la presencia de infecciones como posibles estimulantes del sistema inmune, el tratamiento intensivo (más de 50U/kg de concentrado de factor por más de 5 días continuos) en los primeros 50 días de exposición, como el relacionado con los eventos quirúrgicos 22.
En esta investigación, se analizó la presencia de anticuerpos inhibidores en una cohorte de niños colombianos positivos para las mutaciones del FVIII INV22 e INV1. Se hallaron inhibidores en el 41.6% de los pacientes positivos para la INV22 (5/12) y 33%, para la INV1 (1/3). Estos datos fueron ligeramente superiores a los observados en otros estudios para la inversión de intrón 22 y similares para inversión de intrón 1 17,18,21,24: los hallazgos en población italiana (INV22 25%, INV1 33%), española (INV22 27%, INV1 33%) y alemana (INV22 38%, INV1 33%) y 26.1% en pacientes alemanes con INV1. No obstante, a partir de las evidentes diferencias entre el acerbo genético y los factores medioambientales con las poblaciones mencionadas, es conveniente ampliar la cohorte de estudio con el fin de aclarar o ratificar estos datos.
Según los datos obtenidos en un metaanálisis, se determinó que el OR para desarrollo de inhibidores en pacientes con grandes deleciones fue de 3.6 (IC95%: 2.3-5.7), comparado con la INV22, y de 0.9 (IC95%: 0.6-1.5), comparado con INV1 22. En otro estudio realizado en población norteamericana 25, se estableció un rango de frecuencia de inhibidores en pacientes positivos para la INV22 de 21% a 27%; para mutaciones nonsense, de 25% a 40%; para pequeñas duplicaciones, inserciones y deleciones (incluyendo mutaciones de cambio de marco de lectura), del 10% a 16%, y mutaciones missense, de 5% a 10%. En un trabajo llevado a cabo en población mexicana, la INV22 no fue un factor de riesgo para el desarrollo de inhibidores 16.
Estudios han demostrado que otros factores genéticos diferentes a la mutación causante son una fuente significativa de variación en las frecuencias de inhibidores entre personas que presentan la misma mutación. Esto significa que, más allá del genotipo del FVIII, pueden existir diferencias étnicas en las frecuencias de inhibidores que obedecen a otros factores como algunos haplotipos del factor VIII 15.
Debido a que factores como el número de días de exposición e intensidad del tratamiento han mostrado ser influencias importantes en el riesgo para desarrollar anticuerpos inhibidores en pacientes con hemofilia A severa, se ha postulado que el conocimiento temprano del genotipo de los pacientes podría posibilitar un cambio del plan de tratamiento y así minimizar la exposición a ciertas situaciones en pacientes de alto riesgo, por ejemplo, cirugías electivas y tratamiento intensivo de algunos episodios de sangrado 4,15.
Además, una identificación temprana del genotipo podría facilitar el asesoramiento genético a familias afectadas. En Colombia no existe un esfuerzo organizado a fin de conocer el genotipo propio de la población, a partir de las diferencias propias de su pool genético con el resto del mundo. Esto, con propósitos genéticos y clínicos. De acuerdo con la última encuesta de la Cuenta de Alto Costo y el Ministerio de Salud de Colombia 7, hay 1 325 personas con hemofilia A, 335 tienen inhibidores y cerca del 25% ha desarrollado anticuerpos inhibidores. Según una proyección estadística realizada en población colombiana 25, que expone un estimativo de la carga de la enfermedad genética para el país entre 1996 y 2025, se estima que por cada 10 000 nacidos vivos para el período de 2011 a 2015, se esperan 489 niños con hemofilia y 508 entre 2016 y 2020.
A partir de los datos recogidos en esta investigación, cualquier esfuerzo encaminado a la caracterización genética y epidemiológica de la población de pacientes con hemofilia A parece una tarea con grandes implicaciones y de competencia directa del sistema de salud nacional. Desde la salud pública, el genotipo podría pertenecer a una estrategia con el fin de prevenir anticuerpos inhibidores. Esto podría resultar en un mejoramiento de la calidad de vida de las personas con hemofilia y un ahorro de recursos para estas enfermedades, las cuales son costosas en sí mismas.
Conclusiones
Se detectaron la INV1 e INV22 en un grupo de pacientes con hemofilia A severa a través del uso de metodologías estándar, sencillas y fácilmente reproducibles para esta labor. Se hallaron las inversiones de los intrones 1 y 22 en la mitad de los pacientes evaluados y fueron una herramienta útil en la detección de la INV1 e INV22, las dos mutaciones más frecuentes, lo que posibilita la realización de un tamizaje inicial de la enfermedad.
Se reveló la mutación causante del fenotipo de la enfermedad en el 50% de los pacientes estudiados. Para esta población, en la que no se conoce la mutación asociada a la enfermedad, el paso a seguir es el estudio de las siguientes mutaciones más frecuentes, como las deleciones. Es necesario continuar con el esquema de detección o realizar una investigación del gen completo a fin de caracterizar la población de estudio en su totalidad.
El objetivo de la caracterización de las mutaciones manifestadas en la población de estudio tuvo dos enfoques: primero, desarrollar estrategias que mitiguen el posible desarrollo de inhibidores y segundo, generar una herramienta para el asesoramiento genético de las familias afectadas a través de la detección de portadoras.

