











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
The World Health Organization (WHO) defines spinal cord injury (SCI) as any harmful condition in said tissue, starting from the L1-L2 vertebral level, ending in the conus medullaris and cauda equina. It occurs due to multiple causes, which can be traumatic and non-traumatic, radically transforming the lifestyle of patients. 1
Colombia does not have precise data on the incidence of spinal cord trauma or its repercussions; most studies are based on data from around the world, although they are not accurate either. In 2013, the WHO estimated an annual incidence of 40-80 new cases per million inhabitants, which means that, worldwide, between 250 000-500 000 people suffered some type of SCI, mainly due to traumatic causes, with greater occurrence in young adult men aged 18-32 years and in elderly >65 years. 1,2 In the United States, the incidence of SCI until 2016 was about 54 new cases per million people, while prevalence was estimated at a range of 245 000-330 000 people, with an average age of 42 years. 3
Traumatic injuries correspond to about 90% of the causes of SCI, including those associated with violence, falls, car accidents, and occupational, recreational and sports injuries. Non-traumatic causes of SCI may have an infectious origin or may be related to osteoarthritis and rheumatoid arthritis. Other causes may have a congenital origin such as spina bifida and spinal stenosis or may develop due to a tumor. Certain neurodegenerative pathologies, including those caused by tuberculosis, multiple sclerosis, Alzheimer's disease and Parkinson's disease, are also involved. 1,4,5
SCI morbidity is associated with immunosuppression due to the disruption of the sympathetic innervation of lymphoid tissues caused by neuroendocrine response related to stress and the interruption of the neural pathways that regulate the immune effector functions, which are necessary to control the diffusion of the injury. Consequently, the risk of suffering secondary metabolic, intestinal, urinary and cardiac diseases increases, leading to clinical manifestations such as chronic pain and depression which, in turn, entail a state of permanent disability that severely impacts public health given the high economic and social costs. 1,6-9
Until now, there is no accepted treatment in the world for this condition; pharmacological options are limited and restorative therapies are unsuccessful. Therefore, despite the multiple efforts of the scientific community to elucidate the molecular mechanisms responsible for acute response in SCI and to find new treatments that allow the functional and motor recovery in patients with spinal injury, the outlook is not very encouraging. 5,10
Some therapeutic strategies to deal with SCI aim to establish an enabling microenvironment that favors the activation of endogenous neuroprotection and regeneration mechanisms in the spinal cord to allow axonal projection, promote angiogenesis, control inhibition of the inflammatory cascade, reduce proinflammatory cytokines levels, chemokines and acute phase proteins, down-regulate adhesion molecules expression and attenuate vascular permeability. However, outcomes are usually unsatisfactory, either due to the activation of numerous signaling pathways related to the inhibition of neural growth or the limited regeneration and repair capacity of the central nervous system (CNS). 5,11,12
On the other hand, it is important to consider the therapeutic benefit of controlling inflammation, when it occurs as a physiological response mechanism during the injury. Neuromodulation in said response should be considered, since it is usually related to beneficial conditions for neuroanatomical plasticity and protection against injury or infection, as well as to cytotoxicity states when cytokines concentrations released in the tissue exceed a certain level, altering the homeostasis of the immune system. 11,13,14
The objective of this review is to describe glial cellular response (microglial, astrocytic and oligodendroglial) after a SCI, highlighting the harmful events that determine the progression of tissue damage and, consequently, the difficulty in recovering motor, sensory and autonomic functions in patients. Additionally, an account of inhibitory mechanisms of axonal regeneration, mainly caused by glial activation, favoring the formation of physical barriers in situ that limit timely therapeutic intervention, is also presented.
Materials and methods
A literature search was conducted in the PubMed and Science Direct databases. The following terms were used: SCI, SCI pathophysiology, SCI inflammation, microglia in SCI, glial scar and chondroitin sulfate proteoglycans (CSPG). The 66 most relevant bibliographical references were selected, which corresponded to publications and scientific communications issued in the past 18 years related to physiopathology and glial cellular response after SCI.
Results and discussion
Neuropathological events after traumatic spinal cord injury are classified based on several characteristics. Regarding morphological aspects, SCI may be primary due to physical destruction of membranes and medullary tissue, or secondary due to the events that follow the primary injury, such as hemorrhage, edema, inflammation, reactive gliosis, ischemia, cavitations, excitotoxicity, oxidative stress and neuroglial cell death (Figure 1). Taking into account the biological response, SCI is divided into three phases (Figure 2): acute (0-8 hours after the injury), secondary (2-14 days) and chronic (months to years after the injury). 15-21
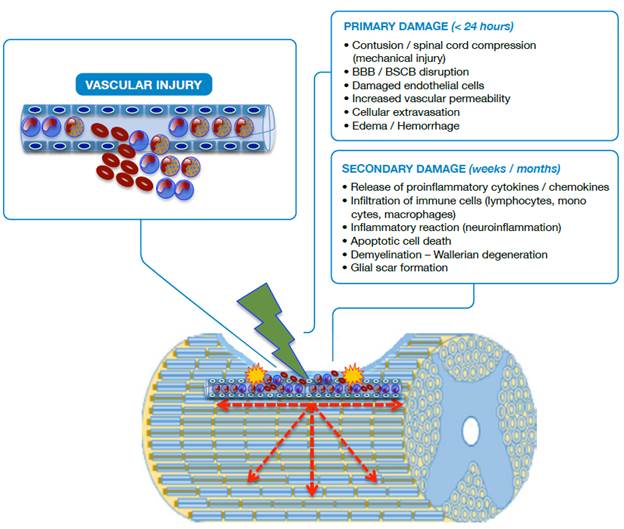
Source: Own elaboration based on the data obtained in the study.
Figure 1 Schematic representation of SCI. BBB: Blood-brain barrier; BSCB: Blood-spinal cord barrier.
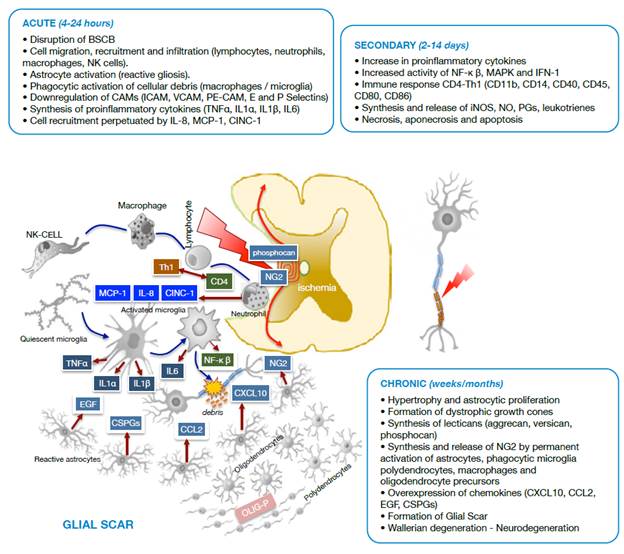
Source: Own elaboration based on the data obtained in the study.
Figure 2 Diagram of cell damage stages after spinal cord trauma. BSCB: Blood-spinal cord barrier; NK cells: Natural killer cells; CAMs: Cell adhesion molecules; ICAM: Intercellular cell adhesion molecules; VCAM: Vascular cell adhesion molecules; PE-CAM: Platelet/endothelial cell adhesion molecules.
In primary damage, it is possible to observe a vascular rupture in the focus of the injury (epicenter) with extravasation of erythrocytes, neutrophils and lymphocytes; disruption of the blood-brain barrier (BBB) and blood-spinal cord barrier (BSCB) is also observed. In secondary damage, there is infiltration of inflammatory cells and the immune system with release of cytokines, chemokines and adhesion molecules that cause the injury to spread in a centrifugal way, increasing its radius. Apoptotic and aponecrotic cell death occurs with involvement of myelin (demyelination), cellular debris, activation of the phagocytic activity of macrophages and barrier formation, known as glial scar, which creates a hostile environment that prevents or delays axonal repair.
The mechanical forces applied to the spinal cord in the primary injury cause the disruption of the endothelial and neuronal membranes, loss of tissue integrity, intraparenchymal hemorrhage, massive cell death, axonal damage and necrotic areas at the site of injury as a consequence of the reduction of blood flow. The first changes in the spinal cord are evident within the first two hours with the expansion of the hemorrhage, the development of vasogenic edema and the presence of diffuse petechiae. 20,22-24
The loss of neuronal cells, especially due to apoptosis, is prevalent between 3-8 hours after the injury, while apoptosis extends beyond 24 hours for glial cells. Although cell loss begins in the epicenter (focus of injury), it progresses rapidly in both directions, ascending and descending with respect to the focus of the injury (Figure 1). About six hours later, the edema reaches the white matter and, at 24 hours, the hemorrhage is significantly extended, the synthesis and the release of proinflammatory mediators at the site of the lesion are increased and the surrounding tissue loses definition. 9,15,25
In the secondary injury, neuropathological processes are associated with accumulation of immune cells such as lymphocytes, monocytes, macrophages and granulocytes 26,27; excitotoxicity; disturbances in the ionic balance; production of free radicals by microglia activation during cellular debris phagocytosis; activation of caspases and calpains, and excessive release of excitatory neurotransmitters and inflammatory mediators. 23,28,29
Vascular changes and blood flow -evident within the first hours after the trauma and persistent for several days and even years after spinal cord injury- lead to apoptotic cell death and axonal demyelination. Likewise, Wallerian degeneration and glial activation with accumulation of astrocyte populations in the periphery and on the surface of the injury continues to give way to the glial scar, a defense mechanism of the nervous system to isolate itself from the noxious influence of the medium 15,18,30 (Figure 2).
In addition, other pathophysiological processes are common and include: increase in blood coagulation; cellular extravasation; chemotaxis and cell adhesion; phagocytosis; angiogenesis; apoptosis; response to hypoxia and reactive oxygen species (ROS), and production and secretion of cytokines, growth factors and cytotoxic amino acids. 5,17,31,32
Cellular response in SCI
During SCI, an active inflammatory process initiates as a defense and tissue repair mechanism, involving a series of cellular components, which respond to the immediate activation of the immune system by creating a physical barrier made up of different cells such as microglia, astrocytes, macrophages, neutrophils and natural killer (NK) cells. On the other hand, the complement system mediates to breakdown the blood-brain barrier, to remodel the extracellular matrix, in the exacerbated activation of astrocytes (reactive gliosis) and in the protection of neurons against cytotoxicity. 24,33
Inflammation, in response to the primary lesion, exacerbates neuronal loss and promotes secondary damage due to the rapid activation of microglia as an intrinsic self-defense line of the CNS and as a resident cell of great importance for immune response because of recruitment, migration and infiltration of neutrophils and perivascular macrophages to the site of the injury, as well as the production of adhesion molecules, metabolites of arachidonic acid and proinflammatory cytokines, both in the acute and chronic state of the injury. 31,32,34,35 This induces a cascade of biochemical events that lead to cell death caused by the mechanisms responsible for neurological deficit such as necrosis, aponecrosis, apoptosis or autophagy (Figure 2). 11,36
During the acute phase, an adverse medium arises, dominated by migration and infiltration of NK cells, neutrophils and macrophages that mediate inflammatory and immune processes through molecules such as MCP-1, CINC-1, IL-8, produced by neutrophils and the Th1 isoform from activated CD4 lymphocytes. It is worth noting that astrocytosis (reactive gliosis) and microglia activate to forms with phagocytic activity and cytokine synthesis (TNFα, IL1α, IL1p and IL6).
During the secondary phase, proinflammatory cytokines increase, as well as the immune-mediated response through CD4 lymphocytes with synthesis and release of active complement molecules (CDs), and the synthesis and tissue release of iNOS, NO, PGs and leukotrienes that exacerbate the inflammatory process. In this stage, the injury radius of cell death of different types increases.
The chronic phase of SCI leads to an adverse environment for tissue repair, involving oligodendrocyte precursors (OLIG-P), polydendrocytes (NG2), activated phagocytic microglia and astroglia that overexpress chemokines. Under these conditions, Wallerian degeneration initiates in the glial scar and neurodegeneration occurs.
As seen in Figure 2, under pathological conditions and in response to molecular factors associated with damage, released after the death of neurons and astrocytes due to the primary injury in the medullary tissue, the resident microglia is activated to initiate the innate immune response, adapted and transformed to other subtypes dedicated to counteract the harmful effects of the injury. 22,35
Although microglia is in a quiescent state in its native form and in normal conditions, thanks to the balance of inhibitory and stimulating signals, it is very sensitive to microenvironmental changes that activate its pattern recognition receptors, against which it begins to have greater motility and phagocytic activity to activate downstream signaling pathways such as the nuclear factor kappa β (NF-κβ) pathways, mitogen-activated protein kinase (MAPK) and interferon 1 (IFN-1). The activation of these pathways promotes the production and release of costimulatory molecules such as surface antigens CD11b, CD14, CD40, CD45, CD80, CD86, proinflammatory cytokines, chemokines, nitric oxide (NO), inducible nitric oxide synthase (iNOS) and arachidonic acid derivatives such as leukotrienes and prostaglandins. 5,22,33,37
Once activated, microglia can remain in that productive state for long periods of time, even at sites distant from the primary injury 24,25,38,39, favoring the release of anti-inflammatory (IL-4, IL-10, TGF-β) and proinflammatory cytokines (TNFα, IL-1, IL-6) that perpetuate inflammation until a chronic condition aggravates by the influx of circulating neutrophils, macrophages, lymphocytes and eosinophils, responsible, as a whole, for greater destruction and tissue loss. 11,22,25,38,40
Clinical and experimental studies have documented the neuroinflammatory process that occurs after SCI in humans and animals. Rice et al. demonstrated elevated levels of IL- 1α, IL-1β,IL-6, TNFα and chemokines within 15 minutes after spinal trauma in an extradural compression murine model. 13 In humans, high serum levels of IL-6, TNFα, IL-1 antagonist receptor (IL-1RA) and antibodies against myelin-associated glycoprotein (GM1) have been identified, the latter in patients with secondary and chronic SCI, between 2 and 52 weeks of evolution. 13,14
Furthermore, the activation of microglia in patients with SCI has been proven to contribute to induction, development and maintenance of neuropathic pain states, mainly due to the production and release of proinflammatory cytokines, chemokines and extracellular proteases. 39 Similarly, microglial activation allows this cell type to adopt antigen-presenting cell functions, as well as other cells similar to macrophages, intervening in innate and adaptive immune response by expressing Toll-like receptors (TLRs), cytokines, chemokines and effector molecules such as CD86 and CD40, which allow the recruitment and activation of other immune cells (lymphocytes, macrophages and NK cells). In addition, activated microglia stimulate the expression of the major histocompatibility complex (MHC) and the response of specific CD4 and CD8 T cells. 35,36,39
Microglial activation marks the beginning of different molecular events that determine the magnitude of the inflammatory response after the trauma, as well as the proliferation, migration and transitory recruitment of other cells at the site of the injury, which perpetuate the release of stimulating cytokines and chemokines, stimulating resident microglia 22,36,41 and causing it to evolve into phagocytic forms. Macrophages, especially neutrophils infiltrated in the injured tissue, have a major role in the inflammatory response, being neutrophils the first cells to migrate to the focus of the injury since the beginning, until reaching its maximum peak at about 24 hours. 22,31
Neutrophils are activated by different stimuli including vascular endothelial damage, phagocytosis and production of proinflammatory substances, and promote the downregulation of intracellular CAM (I-CAM), vascular CAM (V-CAM), platelet/endothelial CAM (PECAM), in addition to E- and P- selectin. The release of IL-8 and chemoattractant cytokines, such as monocyte chemoattractant protein 1 (MCP-1) and cytokine-induced neutrophil chemoattractant (CINC-1), perpetuates the recruitment and activation of more neutrophils thanks to the adhesion to venous capillaries between 6-12 hours after SCI and its migration to the focus of the injury at 24 hours, in order to phagocytose cellular debris. 25,38
At around 48 hours after spinal trauma, the number of neutrophils starts to decline while the number of monocytes increases. They will be later differentiated as macrophages, recruited and activated by TNFα and by the binding of their ligands to the complement receptor 3 (CR-3) and the mannose receptor. On the other hand, macrophages may be observed in the medullar tissue for months and even years, favoring the secretion of glutamate from proinflammatory cytokines (TNFα, IL-1, IL-6), iNOS and prostanoids. 20,25,31,37
Along with microglia, macrophages are necessary for axonal debris phagocytosis and for promoting the production of anti-inflammatory cytokines and molecules that stimulate CNS repair, including the ciliary neurotrophic factor (CNTF), glial cell line-derived neurotrophic factor (GDNF) and insulin-like growth factor 1 (IGF-1). Some neurotrophins, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), are also involved. 20,22,25,31,37
The orchestrated involvement of the inflammatory cells and the immune system is evident in the biological mechanisms that respond to SCI, which are related to both harmful and beneficial processes for tissue repair and regeneration through the regulation of the inflammatory response; the promotion of angiogenesis; the reduction of cytokines, chemokines and acute phase protein levels; the down-regulation in the expression of adhesion molecules; the attenuation of vascular permeability, and the maintenance of homeostasis. 28,42-44
Glial cells, especially astrocytes, play an important role in the functioning of the CNS. They are particularly relevant for controlling the hematoencephalic and hematomedullary barrier, as well as in pH maintenance and ion and neurotransmitter homeostasis in the extracellular space, since they are anchored to the pia mater and the blood vessels from ventricular surfaces by means of GAP junctions. 45 Astrocytes surround 99% of the endothelial cells of BBB and BSCB, which favors their connection with neurons, thus allowing a rapid transfer of substances and chemical information. 28
Inhibitory mechanisms of neuronal regeneration
CNS injuries involve complex cellular and molecular interactions initiated by the recognition of warning signals, which trigger the initial response to the trauma to repair tissue damage. The production of inflammatory molecules and immune surveillance of the CNS allow the entry of specialized cells of the immune system, such as antigen-presenting cells, dendritic cells, macrophages and activated T lymphocytes, to interact with resident cells and trigger the post-traumatic inflammatory response. 42,43 T helper cells (CD4) can change to a different phenotype according to the type of cytokines they release: Th1 produces IL-2, interferon G and TNF, while Th2 produces IL-4, IL-5, IL-6, IL-10 and IL-13. 46
Microglia, macrophages and dendritic cells, which act as antigen-presenting cells after spinal cord injury, trigger an immune response by recruiting leukocytes in the spinal cord and by activating B and T lymphocytes, cytotoxic CD8 and CD4 helpers that increase in number simultaneously as microglia activate, and peripheral macrophages entrance within the first days after the injury. 42,47,48
Moreover, leukocyte infiltration is known to play an important role in injury and spinal repair mechanisms, because it can exacerbate tissue damage and promote myelination and neuronal survival, depending on the microenvironment and the molecular signals that attract them to the site of injury. 48,49
The limitation and gradual reduction in the intrinsic regeneration capacity of mature neurons is also known due to the recruitment of inflammatory cells, reactive astrogliosis and the effect of the production of axonal growth inhibitory molecules from intact polyndendrocytes and oligodendrocytes, like the fibrotic tissue or the glial scar. With this in mind, the non-permissive inhibitory environment, generated by glial cells (astrocytes, oligodendrocytes and microglia), is known to be the main obstacle for CNS regeneration after an injury, contrary to the case of the peripheral nervous system (PNS), where a stimulating environment, product of factors that promote axonal growth, is observed. 12,50-52
Thanks to the contributions of the scientific community, the inhibitory molecular components expressed constitutively or induced in the adult's CNS after a trauma are known today. Microglia, oligodendrocytes and astrocytes participate directly in the establishment of this hostile environment for axonal regeneration due to the production of inhibitors associated with myelin and chondroitin sulfate proteoglycans (CSPGs) present in the glial scar. 51-53
Glial inhibitors of neuronal regeneration
The glial cells of the CNS not only provide structural and physical support, but respond promptly to injuries to provide neural protection through processes related to remyelination, phagocytosis, regulation of neurotransmitters, ionic homeostasis, maintenance of barriers (BBB and BSCB) and production of molecules of the extracellular matrix destined to the formation of the basal lamina and the perineurium. Regarding the injury, these cells respond by forming a glial scar with astrocytes that allow to isolate and protect the intact nervous tissue from future aggressions 52,54 of mature oligodendrocytes and polydendrocytes (NG2 glia) that proliferate in the CNS after neuronal damage and axonal demyelination. 55-57 CNS myelin is formed by oligodendrocyte processes that coil several times around the axon, forming a coating called myelin sheath, interspersed by small unmyelinated or naked segments known as Ranvier nodes. After SCI, oligodendrocytes enter in an early stage of myelin deterioration, unleashing a cascade of secondary events that result in cell death, widespread and dispersed in space, by necrosis, apoptosis and autophagy. This exposes axon regeneration to highly inhibitory molecules, associated with myelin debris (Nogo, MAG, OMgp, ephrin-B3 and transmembrane semaphorin 4D) and to the glial scar formation by reactive astrocytes at the site of the injury. This condition leads to axonal demyelination and spontaneous remyelination generated by new oligodendrocytes that arise from endogenous oligodendrocyte precursor cells in the spinal cord 12,51,52,54,58-60 (Figure 3).
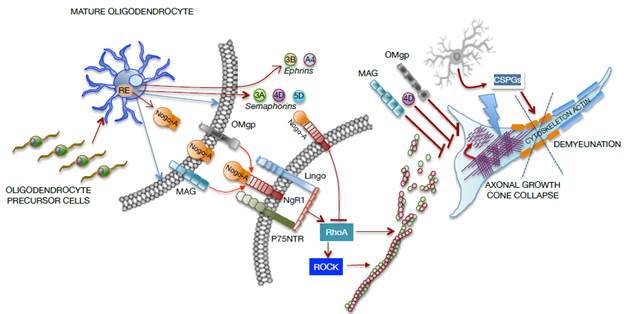
Source: Own elaboration based on the data obtained in the study.
Figure 3 Representative graph of cellular and molecular interaction between mature oligodendrocytes and neurons in a SCI focus. MAG: Myelin-associated glycoprotein; OMgp: Oligodendrocyte myelin glycoprotein.
The axonal growth cone collapses due to the direct effect of molecules from oligodendrocytes, such as myelin-associated glycoprotein (MAG) and oligodendrocyte myelin glycoprotein (OMgp), or due to the formation of the Nogo-A complex and the NgR1- P75NTR-Lingo receptor, which activate RhoA. In turn, the rho-associated protein kinase (ROCK) is activated to prevent the polymerization of actin microtubules, which severely compromise the axonal cytoskeleton. Moreover, oligodendrocytes release transmembrane semaphorins (3A, 4D, 5D) and ephrins (3B, A4), which are involved in growth inhibition and in axonal cone collapse. On the other hand, activated astrocytes (reactants) provide chondroitin sulfate proteoglycans (CSPGs) to the neuronal environment, causing damage to the actin cytoskeleton and demyelination as shown in Figure 3.
a) Myelin-associated inhibitors
Nogo proteins are found in the endoplasmic reticulum and on the cell surface. Nogo-A is the only of its kind found naturally in myelin and is highly expressed in oligodendrocytes, motor neurons and sensory neurons, but not in astrocytes or Schwann cells. 61 Nogo-A inhibits neurites growth by binding its Nogo-66 inhibitor domain to Nogo 1 (NgR1) and PirB receptors, located on the neuronal membrane. 62 This binding forms a complex with LINGO1 and p75 transmembrane proteins in some neurons, which leads to an increase in intracellular calcium and activation of the ROCK pathway. Through RhoA signaling, the actin cytoskeleton is destabilized, resulting in growth cone collapse, which prevents the reestablishment of the injured axons. 51,59,62 On the contrary, blockade of Nogo-A or its receptors promotes plasticity and axonal regeneration of the spinal tracts that survived the injury. 61
Myelin-associated glycoprotein (MAG) is another axonal growth inhibitory protein and, unlike Nogo, is expressed by CNS oligodendrocytes and Schwann cells in the PNS. In the CNS, MAG is located in the periaxonal membrane by the internodal segments of the myelin sheath, whereas in the PNS, MAG is expressed in the paranodal region, in the Schmidt-Lanterman clefts and in other external mesoaxial segments, which allows establishing complexes with axonal surface receptors. 59 It is also known that MAG can be bifunctional due to its powerful inhibitory capacity in neuritic growth under in vitro conditions and for its ability to promote the growth ofyoung neurons in vivo. 51-53,59,63
Oligodendrocyte myelin glycoprotein (OMgp), also described as an inhibitory protein of CNS myelin, is a membrane glycoprotein located near the nodes of Ranvier and expressed in oligodendrocytes and in many types of neurons. It is related to nodal architecture, to growth-cone collapse and to the inhibition of collateral sprouting and neuritic growth. 53,59,64
Nogo, MAG and OMgp show a high interaction affinity with the same receptor complex formed by NgR1, p75NTR and LINGO-1. NgR1 is a neuronal membrane receptor devoid of cytosolic domain, thus forming the complex for signal transduction. The signaling cascade initiated by Nogo, MAG and OMgp results in the activation of RhoA and its effector, Rho-kinase (ROCK). Because of the binding of p75 with the Rho-GDP dissociation inhibitor (Rho-GDI), the organization and dynamics of actin microtubules change and generate signals related to the inhibition of axonal growth by collapsing the growth cone, with the consequent limitation of functional recovery after an injury in the CNS. 51,53,61,63,64
Other components with repulsive axonal growth activity are Semaphorin 3A, 4D and 5, and Ephrins 3B-A4. Semaphorin 4D (Sema 4D) is expressed by mature oligodendrocytes and induces growth-cone collapse; Sema 3A is expressed locally after an SCI; Ephrin 3B is expressed even in postnatal myelinated oligodendrocyte states and acts as a repellent during the projection of the corticospinal tract. 52,55,64,65
b) Inhibitors associated with glial scar formation
The CNS responds to the injury and to the change in the local microenvironment by recruiting microglia, oligondendrocytes precursors, meningeal cells and astrocytes, to isolate the site of injury and minimize the inflammation area and cellular degeneration. However, some astrocyte populations may undergo hypertrophy, change their morphology and gene expression dramatically, and adopt a reactive phenotype (reactive astrogliosis), which may lead to the formation of a glial scar that acts as the main chemicophysical barrier for neural repair by obstructing axonal extension and reconnection, and by inhibiting collateral sprouting of injured axons in the spinal cord. The glial scar, on the other hand, favors the overexpression of proinflammatory cytokines, chemokines (CXCL10, CCL2), endothelial growth factor (EGF) and extracellular matrix molecules such as chondroitin sulfate proteoglycans (CSPGs). 22,55,56,65
CSPGs are a family of molecules formed by a protein nucleus covalently attached to glycosaminoglycans (GAGs) expressed in the CNS, which serve as a guide during the development and modulation of synaptic connections. 4,52,55,60 Some of the CSPGs molecules expressed in the CNS are lecticans, which include aggrecan, versican, neurocan, brevican, phosphocan and the neural/glial antigen 2 (NG2) produced by astrocytes and oligodendrocytes progenitor cells, polydendrocytes, microglia and activated macrophages. In humans, after spinal trauma, NG2 and phosphacan are detectable throughout the glial scar, while neurocan and versican are only found at the epicenter of the injury. 4,52,55
After an injury, the expression of CSPGs increases rapidly due to polydendrocytes and reactive astrocytes, especially in areas near the site of injury, forming an inhibitory regeneration gradient and stimulating the formation of dystrophic growth cones. 4,50,52,60 It is believed that CSPGs inhibit axonal growth due to nonspecific laminin-binding impairment and other extracellular matrix molecules with their transmembrane receptors, and their binding to growth inhibition receptors expressed on the surface of axons. 55 However, it has been determined that the NG2 molecule expressed by polydendrocytes does not inhibit axonal growth when the plasma membrane of the polydendrocyte is intact, thanks to the close synaptic communication established between neuronal dendrites and polydendrocytes. 57 Likewise, CSPGs activate different specific signaling pathways that modulate their inhibitory activity in neural repair, such as those caused by the intracellular activation of RhoA, ROCK, AKT/PKB, mTOR, GSK-3β and protein kinase C, among others. 55
After spinal cord injury, the activation of different cascade down signals that regulate neuronal growth is promoted. The activation of RhoA and the inactivation of Akt by phosphatase and tensin homologue deleted on chromosome 10 (PTEN) are fundamental for activating the signaling pathway produced by the mammalian target of rapamycin (mTOR), for the promotion of protein synthesis involved in axonal regeneration and, consequently, for recovering the locomotor function after the injury. 55,66 In contrast, the activation of the PI3K/Akt/mTOR pathway in reactive astrocytes is involved in the formation of the glial scar during the acute and secondary phases of the SCI. 66
Conclusion
Knowledge and understanding of the neurobiochemical changes that occur after a SCI can contribute to develop new pharmacological therapies that minimize sensory and motor dysfunction in patients with spinal cord injuries who are prone to restoration.
The immune response triggers a cascade of biological events that exacerbate the primary injury and lead to the creation of a hostile environment for neuroregeneration, caused mainly by glial cell activation. The production of proinflammatory and inhibitory molecules of neuronal regeneration is the biggest obstacle for therapeutic intervention, which, in all cases, should seek to establish a favorable microenvironment for the activation of endogenous neuroprotection and regeneration mechanisms in the spinal cord, thus minimizing secondary damage associated with neurological deficit.
The course of SCI is relevant for the type of treatment to be followed regarding basic and applied research, since biochemical events that occur sequentially or simultaneously, as a pathophysiological response to SCI, are the product of the activity of multiple cell lineages and the great diversity of cell signaling molecules and other transient or perpetuating molecules.
Studying cellular and molecular activity of glial cells, such as astrocytes, oligodendrocytes, polydendrocytes and microglia, will allow neurology specialists, and neuroscientists in general, to understand better the process of glial scarring and neuronal death, and to make progress in the knowledge of neuropathology consequent to trauma.

