











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La hipercolesterolemia familiar homocigota (HFHo) es un desorden autosómico dominante. A lo largo de la historia se ha reportado una prevalencia de 1 por cada 1 000 000 habitantes; sin embargo, datos recientes sugieren que esta cifra puede estar entre 1 por cada 300 000 a 160 000 habitantes, o incluso puede ser mayor en poblaciones con características especiales o con mayor frecuencia de consanguinidad 1,2.
Esta patología es causada principalmente por la mutación de ambos alelos del gen del receptor de lipoproteína de baja de densidad (LDLR) 3 y, con menor frecuencia, por mutación de apolipoproteina B100, proproteína convertasa subtilisina/kexina tipo 9 (PCSK-9) o proteína adaptadora dei receptor de lipoproteína de baja densidad (LDL) 1 4.
La HFHo se caracteriza por la presencia de hipercolesterolemia severa (colesterol total >500 mg/dl [>13 mmol/1]), xantomas, xantelasmas y enfermedad cardiovascular severa, que se puede manifestar de forma muy prematura y que en ausencia de tratamiento adecuado ocasiona muerte a edades tempranas 4-6. La actividad residual del LDLR determina la severidad del fenotipo y afecta la respuesta al tratamiento farmacológico 7. Si bien las pruebas genéticas son muy útiles para un diagnóstico certero, un tratamiento oportuno y una modificación del pronóstico, no es usual que sean solicitadas, además el acceso a este recurso es limitado 8.
La mayoría de los pacientes con HFHo responden de manera inadecuada al tratamiento hipolipemiante convencional (estatinas, ezetimibe) 9-12, por lo que se han investigado nuevas intervenciones que han mostrado un beneficio adicional en la reducción de valores de colesterol total y LDL 13; dentro de estos tratamientos se encuentran lomitapida, inhibidor selectivo de la proteína de transferencia microsomal de triglicéridos [MTTP]; mipomersen, inhibidor de apoB-100, y Evolocumab y Alirocumab, inhibidor de PCSK-9 y aféresis.
Se presenta el caso de dos hermanas con diagnóstico clínico, bioquímico y molecular de HFHo, lo cual permitió enfocar y optimizar el manejo farmacológico hipolipemiante y lograr una reducción significativa en los niveles séricos de colesterol total y LDL sin nuevos eventos cardiovasculares.
Presentación de los casos
Pacientes de sexo femenino de 43 (paciente 1) y 56 años (paciente 2), ambas con antecedente de hipercolesterolemia severa, hipertensión arterial y enfermedad coronaria multivaso, por lo que requirieron intervención coronaria percutánea y quirúrgica con anticipación. Al examen físico tenían xantelasmas y xantomas múltiples en las extremidades (Figuras 1 y 2). En cuanto a los antecedentes familiares, tenían un hermano con hipercolesterolemia familiar que falleció a los 33 años de edad por muerte súbita. La paciente 1 recibía losartán, metoprolol, hidroclorotiazida, clopidogrel y ácido acetilsalicílico y la paciente 2 recibía enalapril, carvedilol y ácido acetilsalicílico; ambas recibían rosuvastatina (40 mg/día) y ezetimiba (10 mg/día). A pesar del tratamiento farmacológico hipolipemiante intensivo, no se habían logrado metas de control lipídico (colesterol total y LDL).
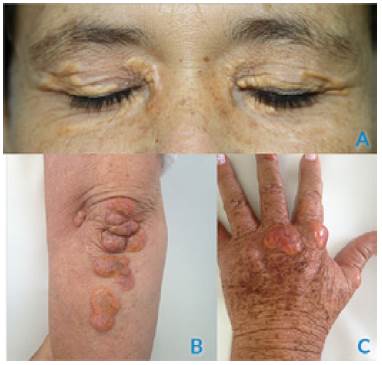
Fuente: Documento obtenido durante la realización del estudio.
Figura 1 Paciente 1. A) Xantelasmas; B) xantomas en codo; C) xantomas en mano.
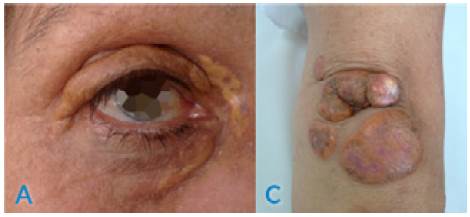
Fuente: Documento obtenido durante la realización del estudio.
Figura 2 Paciente 2. A) Arco corneal y xantelasmas; B) xantomas en codo.
Por lo anterior, en ambos casos se realiza secuenciación completa del gen LDLR que muestra deleción en homocigosis de los exones 4 al 6 (p.Pro 105_Gly314delinsArg), sin cambios en el marco de lectura o "in frame". Estos exones codifican el dominio de unión al ligando de LDLr que facilita la interacción con lipoproteínas, por lo tanto, su deleción resulta en una pérdida de unión de LDL a LDLr (Figura 1), lo cual se refleja en el aumento de la concentración plasmática de dicha lipoproteína. Esta mutación encontrada es de alelo defectuoso, es decir, existe función residual.
El mecanismo molecular que conduce a la deleción de estos exones se conoce como "replication con ruptura inducida mediada por microhomologia" y se basa en la recombinación homologa entre dos secuencias Alu, en este caso específico entre AluSq en el intrón 3 y AluSc en el intrón 6.
Teniendo en cuenta los hallazgos de las pruebas diagnósticas y la pobre respuesta al tratamiento hipolipemiante convencional, se decidió adicionar un inhibidor de la MTTP (lomitapida). En ambos casos la dosis inicial del medicamento fue 5 mg/día durante 1 mes y luego 10 mg/día. Por problemas con la aseguradora de las pacientes, el medicamento no fue suministrado durante un periodo de tiempo en el que el colesterol tuvo de nuevo un incremento importante (Figuras 3 y 4). Las dos pacientes tuvieron buena tolerancia al medicamento, no presentaron efectos adversos importantes y las enzimas hepáticas se mantuvieron dentro de rangos normales.
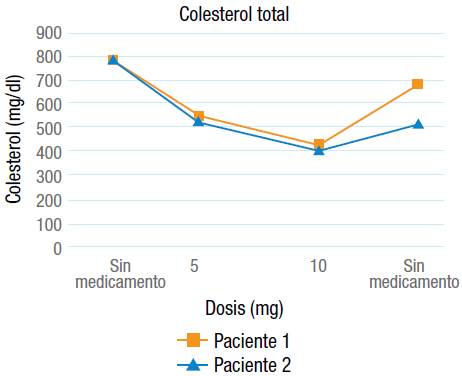
Fuente: Elaboración propia.
Figura 3 Niveles séricos de colesterol total relacionado con la dosis de lomitapida (adicionado al tratamiento hipolipemiante convencional).
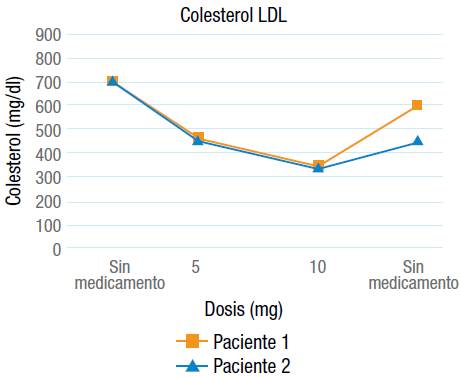
Discusión
El diagnóstico diferencial de la HPHo incluye hiperlipidemia familiar combinada, hiperlipoproteinemia tipo III (disbetalipoproteinemia), xantomatosis cerebrotendinosa, sitosterolemia e hipercolesterolemia familiar heterocigota grave 5. Aunque las características de cada patología pueden sugerir el diagnóstico clínico, en muchos casos es necesaria la realización de pruebas moleculares para confirmarlo 14.
Las características moleculares de pacientes con HFHo se han asociado con diferencias en el fenotipo, respuesta al tratamiento y riesgo de enfermedad coronaria temprana, por esto, la evaluación genética y los estudios moleculares son útiles para estimar la severidad clínica y el pronóstico de la enfermedad 7. Con respecto a la mutación de alelo defectuoso como la de las pacientes presentadas, se han mostrado diferencias en cuanto a las características clínicas en comparación con las mutaciones de alelo nulo; en estas últimas se ha reportado inicio de enfermedad coronaria a edades más tempranas más estenosis valvular aórtica y necesidad de trasplante hepático, además valores de colesterol sérico significativamente mayores respecto a los pacientes que tienen mutaciones de alelo defectuoso 15.
En las pacientes estudiadas se inició tratamiento con un inhibidor selectivo de la proteína de transferencia microsomal de triglicéridos [MTTP]. Aunque existen otras opciones terapéuticas que podrían haberse considerado, los inhibidores de Apo B-100 no están disponibles en la actualidad y los inhibidores de PCSK-9 son menos efectivos en pacientes con HFHo debido a que estos fármacos actúan incrementando la expresión del LDLR que ya está afectada en HFHo 16. Por otro lado, el uso de aféresis como manejo complementario no se consideró en estas pacientes debido a la excelente respuesta farmacológica observada hasta ahora. Es importante mencionar que este procedimiento no está disponible en la mayoría de instituciones en Colombia 17.
Con base en los resultados de las pruebas moleculares, se decidió iniciar tratamiento con un inhibidor selectivo de la MTTP, el cual ha demostrado eficacia como monoterapia o cuando se adiciona al manejo hipolipemiante convencional para el tratamiento de la HFHo 3,18. En los dos casos se inició manejo con 5 mg/día y luego 10 mg/día, además del tratamiento hipolipemiante convencional. En la décima semana de tratamiento se observó una reducción promedio de 49% para el colesterol total y LDL (Figuras 3 y 4). A pesar de ser un corto tiempo de tratamiento, hubo porcentajes de disminución en los niveles séricos de colesterol (total y LDL) cercanos a lo reportado en la literatura 14.
Debido a la orientación dada por el estudio molecular, se logró optimizar el manejo de las pacientes, lo cual tuvo beneficios en las concentraciones de colesterol, que como se ha mostrado en estudios retrospectivos impacta de forma positiva en la morbimortalidad de los pacientes con HFHo 15,16,19.
Conclusión
Las pruebas genéticas son una herramienta importante para el diagnóstico de la HFHo, por lo tanto el acceso a ellas podría impactar de manera significativa el pronóstico de los pacientes con esta patología, pues permite adicionar nuevas terapias hipolipemiantes que a su vez permiten lograr disminución importante en el nivel de colesterol total y LDL.
Consideraciones éticas
En el presente reporte de caso se contó con consentimiento informado por parte de las pacientes.

