











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Henoch-Schönlein purpura (HSP) is the most common form of vasculitis in childhood. It is an immunoglobulin A (IgA)-mediated disorder characterized by palpable small-vessel vasculitis. About 90% of HSP cases occur during the first 10 years of life, and the most common age range is 3-5 years.1,2 Lesions are usually observed on the lower limbs and sometimes on the upper limbs, face and chest. Moreover, most patients have petechial or purpuric lesions or a combination of both; also, some individuals have bull's eye lesions consisting of concentric rings surrounded by pale and hemorrhagic rings.
The most common gastrointestinal symptoms in HSP are abdominal pain, vomiting, diarrhea, and paralytic ileus. However, non-erosive arthritis can also occur without permanent deformity in the ankles, knees, and feet. 2 As for skin lesions, blisters are the least frequent and are found in about 5% of cases: Saulsbury3 found 2 patients with hemorrhagic bullous lesions in a series of 100 patients.
This case report describes an atypical presentation of a skin condition in a patient treated at a tertiary pediatric hospital in Bogotá D.C., Colombia.
Case presentation
A five-year-old male patient, with a history of bronchiolitis and pneumonia, was taken by his parents to a primary care center due to arthralgia in hands, feet, and knees associated with the appearance of painful and erythematous lesions in the skin of the lower limbs; these symptoms appeared three days before consultation. The child was diagnosed with HSP. Laboratory tests were performed, including urinalysis, renal function, and urinary tract ultrasound, all of which were normal and therefore treated on an outpatient basis.
Two days after discharge, the parents took the child once again to the medical center due to a complication of his symptoms, so he was referred to a tertiary care center. The skin lesions turned into multiple phlyctenae containing translucent blood-like fluid, with variable sizes and located exclusively in the lower limbs. The physical examination revealed stable vital signs and grade 2 edema in the lower limbs accompanied by multiple palpable purpuric lesions extending to the buttocks; in addition, some of these lesions appeared in isolation in the upper limbs. Many of the skin lesions joined together and formed phlyctenae with serohematic fluid (Figure 1), mainly in the feet and legs, which generated pain on palpation and as a result of mobilization of the lower limb joints.
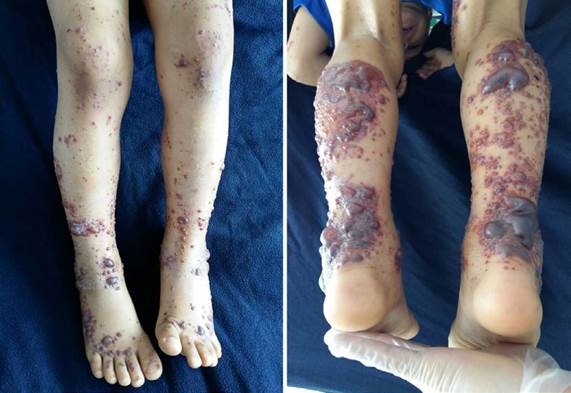
Source: Document obtained during the study.
Figure 1 Purpuric lesions and phlyctenae on the lower limbs.
After the physical examination, the diagnosis of HSP was reconfirmed, and the patient was hospitalized given his symptoms, mainly a severe skin condition; laboratory tests were performed, and intravenous analgesia with opioids was adjusted. The tests (Table 1) revealed low-grade proteinuria with a urine protein/creatinine ratio of 0.43, urea nitrogen of 25.8 mg/dL, and creatinine of 0.45 mg/dL, showing a BUN/creatinine ratio of 57.3. Acute kidney injury in pregnancy secondary to dehydration was considered, so intravenous fluids were adjusted, and kidney function tests and abdominal ultrasound were requested.
Table 1 Lab test results.
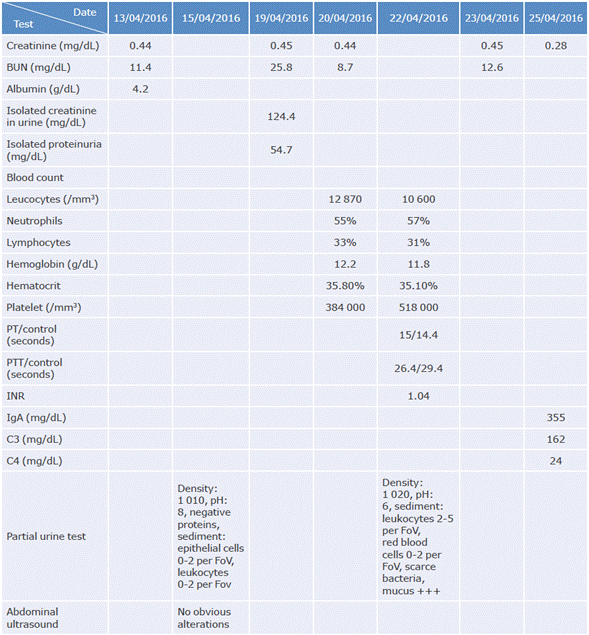
BUN: blood urea nitrogen; PT: prothrombin time; PTT: thromboplastin time; INR: international normalized ratio; IgA: immunoglobulin A; C3: complement component 3; C4: complement component 4; FoV: field of view of microscope.
Source: Own elaboration.
Kidney function test, glomerular filtration rate, kidney ultrasound, complement test, and coagulation times were normal during hospitalization; only a discrete elevation in IgA levels was found. The patient was assessed by the plastic surgery and dermatology services, which agreed with the HSP diagnosis. Given the large skin involvement, a skin biopsy was performed, finding neutrophilic intradermal vesicles that involved the dermis; immunofluorescence was not reported in the pathology results.
The patient recovered satisfactorily and received support treatment with a vaseline gauze dressing over the bullous lesions, analgesia, and intravenous hydration for the pain. Subsequent kidney function tests were normal, so he was discharged.
Discussion
Among systemic vasculitides, HSP is the most frequent in the pediatric age, but its diagnosis is questioned when the clinical presentation does not match the classic expression of the disease.1,2,4 HSP is an IgA-mediated systemic small blood vessel vasculitis and may result from a response of immune complexes to various antigenic stimuli in susceptible individuals.1
The most common symptoms of HSP are purpura, arthritis, abdominal pain, gastrointestinal bleeding, and nephritis;2 only 2% of patients develop bullous hemorrhagic lesions.1
According to González et al.,5 the incidence of this pathology is 135 cases per million inhabitants in the population under 14 years of age, being 100 times lower in adults.5 In turn, Rabelo et al.6 reported an annual incidence that ranges from 13.5 to 18 cases per 100 000 inhabitants for the year 2000 in Brazil.
The etiology of HSP is not clear, but some association has been found with a history of group A beta-hemolytic streptococcal infections, Salmonella spp, Bartonella spp, Yersinia or Mycoplasma spp. Other germs related to this disease are the hepatitis B virus, human immunodeficiency virus, Epstein-Barr virus, Coxsackie virus, chickenpox virus, and adenovirus. The onset of HSP has also been associated with the use of some antibiotics such as penicillin or macrolides, or angiotensin-convert-ing enzyme inhibitors, angiotensin II receptor blockers, and non-steroidal anti-inflammatory drugs.3,7
HSP is a leukocytoclastic vasculitis that sometimes presents with increased IgA levels; it may be caused by microorganisms that share an antigenic determinant with small-caliber blood vessel endothelial cells. In this sense, after pathogen invasion, the antigen-specific IgA antibodies cross-react with the antigens of some pathogens and are deposited in the endothelial cells, mainly in the skin and kidney, although they can also be deposited in the joints and the intestine, causing the symptoms and signs of the disease. However, this is only a theory, since no association has yet been found between a specific antigen and the development of HSP.
Another hypothesis that could explain the presence of HSP is related to the alteration of oligosaccharide glycosylation in the hinge region of IgA1, which generates abnormal IgA1 levels that are not completely catabolized in the liver and are deposited in various tissues, such as in the kidney mesangium. After this process, the complement system is activated alternately, generating damages in the endothelial lining; at this point, an increase in IgA and C3 complement in the endothelial cells of the skin and kidney mesangium is evident.
The clinical signs of HSP occur in the organs where the largest deposits of immunoglobulin are found. Therefore, four main clinical manifestations have been established: 1) Purpuric skin lesions, which are palpable and predominantly present in the lower limbs but can be also be found in other body areas to a lesser extent; 1,8,9 2) arthralgia or arthritis, which are generally oligoarticular (predominantly in the ankles, feet, knees, and hips and to a lesser extent in the upper limbs) and migratory; 3) abdominal pain, which may be related to gastrointestinal bleeding; and 4) kidney impairment, which may lead to acute kidney failure in severe cases. 3,10
Given the presence of multiple etiological agents and non-specific prodromes, it is necessary to make a correct initial diagnosis of HSP, since kidney and gastrointestinal involvement may exist in 40% and 33% of cases, respectively, although most clinical presentations are found in the skin. It is worth noting that 2% of patients with skin lesions may develop bullous or blistering lesions without a clear trigger, as in the present case, in which the patient presented with typical lower limb HSP lesions that progressed to bullous lesions.
Despite the self-limiting course of the disease, significant sequelae and risk of short- and long-term complications can be observed, such as nephritis, kidney failure, massive gastrointestinal bleeding, and relapse.3 Saulsbury3 also reported alterations in some kidney function tests such as urine protein/creatinine and BUN/creatinine ratio, which, in this patient, were related to a prerenal problem caused by dehydration and not to a frank kidney injury, since the laboratory tests, once dehydration was corrected, were normal during hospital stay until discharge.
Since the bullous presentation of HSP is rare, it is essential to differentiate it from other types of vasculitis and other skin pathologies since their management is different, and the sequelae and complications may be severe. Based on this, in the present case, the diagnosis was confirmed by the dermatology service, which ruled out other possible differential diagnoses.
According to the experience and the literature reviewed by Chen et al.8 and Liu et al.,9 severe cutaneous presentations are associated with a very low probability of kidney or gastrointestinal involvement, as is the case reported here. Conversely, mild skin involvement can lead to significant kidney or gastrointestinal involvement, making a correct diagnosis necessary, as evidenced by Saulsbury3 in his case series.
Conclusions
The available literature suggests that the cutaneous presentation of bullous HSP is rare in pediatrics and is not always related to kidney and/or gastrointestinal involvement, as is the case of the classic variant. However, the function of these systems should be monitored regardless of the severity of this vasculitis on the skin, as life-threatening complications could arise in the short or medium term. Therefore, outpatient follow-up should be performed in the first months after the diagnosis, requesting laboratory tests such as complete blood count, urinalysis, and kidney function and keeping adequate blood pressure, height, and weight levels to evaluate the evolution and status of the patient.

