











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Precocious puberty is defined as the appearance of secondary sexual characteristics before the age of 8 in girls and 9 in boys.1-3 It is classified into central precocious puberty (CPP), which is caused by the early activation of the hypothalamic-pituitary-gonadal axis, and peripheral precocious puberty (PPP), which occurs when there is no activation of the gonadotropic axis.
PPP is characterized by the production of sex hormones that may be caused by genetic factors (congenital adrenal hyperplasia (CAH), McCune-Albright syndrome, mutation of the DAX1 gene and familial testotoxicosis), or acquired factors (functional ovarian cysts, gonadal or adrenal tumors, (3-hCG-secreting tumor and consumption of exogenous sex steroids).2,4 The following is the case of a girl diagnosed with PPP secondary to non-classic CAH.
Case presentation
A 4 1/2-year-old female patient was assessed by the pediatric endocrinology service due to the appearance of pubic hair six months before the consultation, without thelarche or acne. Her parents were first-degree cousins, and, given their heights, the mid-parental height would be 150cm (less than 2 standard deviations).
At birth, her weight was 3340g, length 50cm, and head circumference of 34cm. She presented early symptomatic neonatal hypoglycemia that required intravenous administration of dextrose, and jaundice that was treated with phototherapy. The patient also presented left spastic hemiplegia secondary to left lateral ventriculomegaly, which had been identified on prenatal ultrasound.
The physical examination revealed: height: 105.3 cm (0.5 standard deviations, according to the growth charts from the Centers for Disease Control and Prevention, USA); weight: 17 kg; body mass index: 15.4 kg/m2; bone age: 6 years according to Greulich and Pyle method; normal blood pressure; absence of goiter, abdominal masses and acne; Tanner breast 1 and pubic 2; and female genitals without clitoromegaly.
Based on these findings, the patient was diagnosed with PPP secondary to non-classic CAH. An adrenocorticotropic hormone (ACTH) stimulation test was requested (Table 1), finding normal renin and electrolyte levels in blood, as well as high levels of 17-hydroxyprogester-one (17-OHP), which are clinical signs of CAH.
Table 1 Laboratory tests.
| Test | Result | Reference values | ||
| ACTH Test | 17-hydroxyprogesterone | Pre-stimulation | 0.7 ng/mL | <2 ng/mL |
| Post-stimulation | 25.1 ng/mL | <15 ng/mL | ||
| Cortisol | Pre-stimulation | 212.1 nmol/L | 276-552 nmol/L | |
| Post-Stimulation | 921.9 nmol/L | >848.4 nmol/L | ||
| Free testosterone | 1.3 pg/mL | <0.5 pg/mL | ||
| DHEA | 947.3 ng/mL | 0.32-5.84 ng/mL | ||
| Delta 4-androstenedione | 2.35 ng/mL | <0.5 ng/mL | ||
ACTH: adrenocorticotropic hormone; DHEA: dehydroepiandrosterone.
Source: Own elaboration.
In the absence of hydrocortisone, prednisolone was administered at a dose equivalent to 15 mg/m2/day of hydrocortisone, thus lowering the levels of dehydroepiandrosterone (DHEA), androstenedione, testosterone, and 17-OHP. During a follow-up consultation, her chronological age (9 years) was consistent with her bone age and height in less than 2 standard deviations (Figure 1), which coincided with her mid-parental height.
Discussion
The onset of puberty, which may be early, in clinical terms, occurs when the breast bud appears in girls and when the testicular volume is >4mL in boys.5 In CPP, the appearance of secondary sexual characteristics occurs sequentially, contrary to what happens in PPP.
The appearance of pubic hair before age 7 in white girls, before age 6 in African American girls, and before age 9 in boys is defined as early puberty.6,7 Also, the presence of pubic hair in the absence of breast growth in girls suggests adrenal and ovarian disorders or exposure to androgens.5 In the reported patient, pubarche, as the first finding of puberty added to the advanced bone age and the lack of concordance of height compared to mid-parental height, indicated the presence of PPP.
One of the causes of PPP is CAH, an autosomal recessive disease characterized by an alteration of adrenal steroidogenesis that leads to a decrease in the synthesis of cortisol and aldosterone. Such decrease generates negative feedback in the pituitary gland with a consequent ACTH overproduction and subsequent stimulation of the adrenal gland, which in turn causes hyperplasia.8
About 95% of CAH cases are caused by a 21-hydrox-ylase deficiency8,9 due to the mutation of the CYP21A2 gene. There are 2 forms of presentation: classic and non-classic. The prevalence of the former is 1 case per 16 000 births, while the prevalence of the latter is 1 case per 1 000 births. 6,10,11
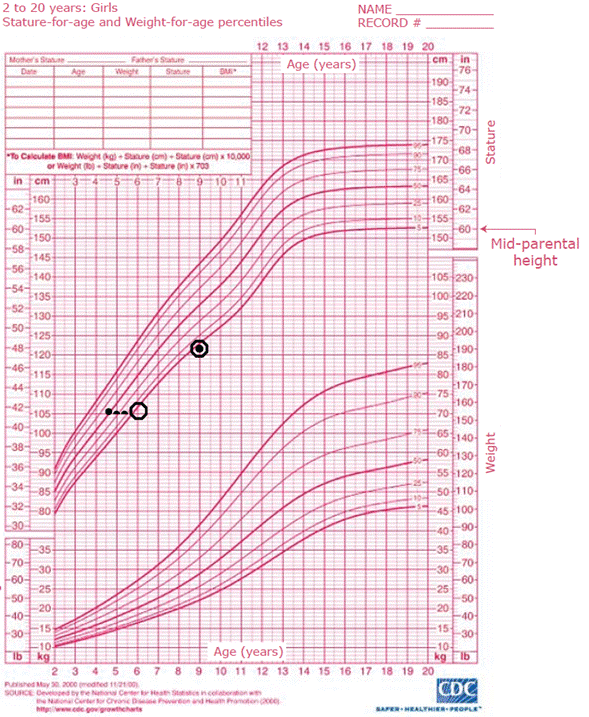
• Height O Bone age. Source: Own elaboration.
Figure 1 Centers for Disease Control and Prevention growth chart plotted according to chronological age
Most patients with non-classic CAH are asymptomatic or have mild manifestations that may be expressed as precocious puberty, hirsutism, acne, clitoral hypertrophy, menstrual irregularities, or polycystic ovary.8,12,13 In this regard, Moral et al.14 conducted a multicenter study in 220 girls with non-classic CAH and found that 92% of the girls under 10 years of age had early puberty.
Hypoglycemia may also be present in non-classic CAH due to Cortisol deficiency, a counter-regulatory hormone that increases blood glucose levels through gluconeogenesisand glycogenosis. 15,16 In the reported case, it was present in the neonatal stage, a time of adaptation with high metabolic demands that include an increase in counter-regulatory hormones, which went unnoticed, as in many cases.
The diagnosis of non-classic CAH is made by determining the concentrations of 17-OHP, a metabolite that builds up as a result of the steroidogenesis disruption caused by the 21-hydroxylase deficiency. Therefore, Speiser et al.,17 in their 2010 consensus, suggest measuring 17-OHP in blood in the morning and performing a full adrenocortical profile after an ACTH stimulation test to differentiate 21-hydroxylase deficiency from other enzyme defects, or in case of doubtful diagnosis.
For non-classic neonatal CAH screening, a blood sample on filter paper is collected by pricking the baby's heel between the second and fourth day of life, which is tested to obtain 17-OHP levels using mainly immunoassay techniques. The result is considered abnormal when levels are above the 97th percentile for age. 6,8,11
Unlike classic CAH, random 17-OHP levels in non-classic CAH may be in the normal range; therefore, the ACTH stimulation test is the gold standard for diagnosis18,19. It's considered positive when pre-stimulation levels are >5 ng/mL (15nmol/L) and post-stimulation levels are >15 ng/mL (45 nmol/L). 8,17,18 In the reported patient, the baseline 17-OHP level was normal, and was only elevated on the ACTH test, which indicated non-classic CAH. Testosterone and delta 4-androstenedione values were higher than expected for the pre-pubertal stage, which contributed to the diagnosis of CAH and its corresponding decrease after treatment.
Since CAH is an autosomal recessive disease, some research12,13,17 suggests that genetic studies should always confirm the CYP21A2 gene mutation and that phenotype-genotype correlation should be made to provide genetic counseling for the family. However, in the present case, it was not possible to carry out such studies and, therefore, counseling was not provided due to problems related to the patient's social security coverage.
Glucocorticoids are administered for the treatment of non-classic CAH in patients with accelerated bone age, virilization, and premature or rapid progression pubarche.9,17,18 Some of the objectives of this treatment in girls are achieving adequate growth rate and the proper onset of puberty, as well as avoiding accelerated skeletal maturation, the reduction of the expected mid-parental height, and psychological alterations. In adolescents, the treatment aims to avoid irregular menstrual cycles, hirsutism, and acne. Likewise, long-term corticosteroid use can prevent frequent situations caused by this pathology, such as infertility, abortions, fetal death, psychiatric problems, decreased bone density, obesity, dyslipidemia, insulin resistance, hypertension, diabetes, among others.18,19
It is worth mentioning that the growth rate increases in precocious puberty. Therefore, one of the best ways to assess whether the treatment is appropriate is by evaluating this aspect. In the case presented here, the girl initially had a difference of more than 2 standard deviations from the mid-parental height, but upon receiving treatment, she returned to her normal growth rate and her bone age did not progress. 17-OHP and delta 4-androstenedione values should be reviewed during follow-up to confirm if normal levels have been achieved, as is the case of this patient.
Conclusions
Non-classic CAH is the most common cause of PPP. Since this type of hyperplasia may be asymptomatic during the first days or years of life, this diagnosis should be suspected when there is early puberty, increased growth rate, and advanced bone age. Early treatment of CAH helps avoid loss of final genetic height and prevent cardiometabolic diseases in adulthood.

