











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introduction
The prevalence of thrombotic diseases in children has increased as the molecular processes involved in their pathophysiology have been better characterized. Several inherited disorders are described as triggers of these diseases, including deficiencies of the fibrinolytic system and anticoagulation factors, metabolic alterations, hemoglobinopathies, platelet hyperaggregation and various acquired autoimmune diseases, endocrine disorders, neoplastic diseases, infections, and chronic diseases (kidney, liver, among others.)1
After the discovery of platelets and the description of their physiological function, research on these cells has focused predominantly on bleeding disorders2 and they are now known to have multiple biological functions involved in hemorrhagic and thrombotic phenomena;3 however, it should be noted that platelet-associated thrombotic events are considered rare.4
Sticky platelet syndrome (SPS) is a familial thrombophilic condition of autosomal dominant inheritance characterized by an increase in platelet aggregation leading to arterial and venous thrombosis. SPS was first described by Holliday et al.5 at the Ninth International Joint Conference on Stroke and Cerebral Circulation in 1983, and later, in 1986, Rubenfire et al.6 published the first study describing patients with cardiovascular thrombotic events related to platelet hyperaggregation.
Then, in 1988, in a study describing cerebrovascular thrombotic events, Mammen et al.7 established the first two criteria for identifying SPS in venous blood samples, using sodium citrate as anticoagulant and three concentrations of two agonists: 1) tests with adenosine diphosphate (ADP) at concentrations of 2.34uM, 1.17uM and 0.58uM, with which aggregation ranges of 7.5-55%, 2-36% and 0-12%, respectively, were obtained in control subjects, and 2) tests with epinephrine at concentrations of 11uM, 1.1uM and 0.55uM, with which aggregation ranges of 39-80%, 15-27% and 9-20%, respectively, were obtained in control subjects. Furthermore, the researchers found that in patients with SPS, these values were significantly increased. Subsequently, Mammen8 described various thrombotic disorders related to platelet hyperaggregation in adults and in some pediatric cases; in addition, the diagnostic parameters of the SPS described above were confirmed in this study.
Finally, Bick9 published a study of 153 adults residing in Dallas, USA, who presented with thrombotic events, in which they found that 33% of participants met the diagnostic criteria for SPS. Based on the characteristics of these patients, the author recognized three types of the disease:
Type I: when hyperaggregation is observed when using epinephrine and ADP.
Type II: when hyperaggregation occurs only with epinephrine.
Type III: when hyperaggregation occurs only with ADP.
Bick9also established that the diagnosis is suggestive of SPS when, besides thrombosis, there is hyperaggregation along with a concentration of ADP or epinephrine higher than the normal ranges previously described in only one of the tests and that this is confirmed with one of the following possibilities: a) history of thrombotic disorder and presence of hyperaggregation with two concentrations of one of the two agonist reagents (ADP or epinephrine); b) history of thrombotic events and platelet hyperaggregation with one concentration of the two agonist reagents; and c) history of previous thrombosis and presence of hyperaggregation, with the same concentration of one of the agonists, that persists when the test is repeated.
The first case of SPS reported in Colombia in pediatrics was described in 2002 by Beltran et al.,10 who also stated that this entity is an important cause of thrombotic disorders during childhood. Later, also in Colombia, Velasquez et al.11 published a series of cases of pregnant women diagnosed with SPS, in which they concluded that it is an unknown disorder despite being one of the most frequent causes of thrombotic events. Similarly, studies related to platelet hyperaggregation syndromes have been carried out in several countries, finding that the same inherited phenotype associated with SPS can be observed in certain metabolic conditions such as diabetes mellitus and atherosclerosis, or in some inflammatory and infectious diseases.12-15
Other research, such as Kubisz et al.16 suggest that some thrombotic events without known etiology may be caused by SPS. Moreover, genetic studies have reported mutations and polymorphisms in the genes of the glycoprotein VI found in the platelet membrane, integrin beta 3 sub-unit, suggesting that variability in these genes may be related to SPS, although no significant differences have been found between patients with and without the disease.17,18
Recent studies relate different mechanisms of hemostasis to activity-dependent intracellular calcium initiated by cell membrane receptors, which are activated by intracellular signals and modulated by calcium signaling receptors located on the membrane of the endoplasmic reticulum, with positive or negative feedback on receptors located on the cell membrane.19,20
Furthermore, Barros-Garcia et al.,21 after analyzing the exome in an index case with SPS, identified a molecular alteration consisting of the deletion of 5 nucleotides of the ORAI1 gene (located in the long arm of chromosome 12), which resulted in an erroneous reading in protein synthesis. These authors also described that the protein encoded by this gene is the calcium channel subunit of the membrane activated by the calcium sensor STIM1 when calcium reserves are depleted in the cell.21
Similarly, although progress has been made in the understanding of platelet hyperaggregation in pediatric patients, many aspects remain to be clarified. Next-generation sequencing of the exome could be useful for this purpose since it has been possible to identify strategies to detect the genetic causes of different hereditary diseases.
Exome sequencing is a test used to identify DNA variants that cause genetic disease. It has been established that most known variants that cause disease occur within 1% of the genome containing exons and at the cleavage and splice junctions; therefore, studying this part of the genome is an effective and powerful clinical diagnostic tool in personalized medicine.22-24 Thus, the objectives of this study were to identify common mutations by sequencing the exome in two patients from the same family diagnosed with SPS and, consequently, contribute to the molecular study of this disease.
Materials and methods
Descriptive study in which exome sequencing was performed on a pediatric patient (10-year-old index case at the time of the study) and in one of his first-degree adult relatives (mother). Both the index case and two of his relatives (mother and sister) had a history of thrombotic disease and had been diagnosed with SPS.
Description of index case
After reviewing the medical records of patients diagnosed with SPS treated at the Fundación Hospital Pediátrico de la Misericordia (HOMI) in Bogotá, Colombia, from 2001 until the time of the study, a 10-year-old male patient, with no history other than a thrombosis of cerebral artery territory was selected as the index case. In his clinical history, it was found that one of his relatives had a history of a transient ischemic attack and migraine and that both patients had a history of platelet hyperaggregation with ADP and epinephrine.
Both individuals were diagnosed with SPS by platelet aggregometry and using the guidelines proposed by Mammen8 and Bick.9 In addition, to select the cases, it was confirmed that the tests had been carried out while the patients were not receiving antiplatelet treatment and three months after the acute thrombotic event had occurred in each case.
Moreover, a normalization of platelet aggregation curves with ADP and epinephrine using acetyl salicylic acid as a treatment was documented. Likewise, other possible causes of SPS related to congenital thrombophilia were ruled out through specific studies that yielded negative results.
In both patients, blood samples were taken at the Molecular Biology Laboratory of the Universidad Nacional de Colombia, where they were processed and stored, and DNA was extracted.
DNA sample separation
Blood samples were obtained by venipuncture in a 5mL Vacutainer® tube with EDTA anticoagulant and stored at -80°C until processing. DNA was extracted using the PureLink™ Genomic DNA kit, following the manufacturer's instructions (Invitrogen), and from mononuclear cells isolated by sucrose-gradient centrifugation using LSM (Lymphocyte Separation Medium) reagent.
DNA concentration was determined using the Thermo Scientific™ NanoDrop 2000 spectrophotometer, while the purity and quality of these DNA samples were determined by electrophoresis and through the 260/280 to 260/230 absorbance ratio, which ranged from 1.8 to 2.0.
For processing on the sequencing platform, the DNA integrity level of the samples was verified using 0.8% agarose gels or 2200 TapeStation (Agilent Technologies) with Genomic DNA ScreenTape assay (Agilent Technologies) chips.
Exome analysis
Exome sequencing of patient samples was performed at the Cardiovascular Genetics Laboratory of the Instituto de Investigación Sanitaria del Complexo Hospitalario Universitario de Santiago de Compostela, Spain, using the SureSelect Clinical Research Exome V2 software of Agilent.
Samples were processed to prepare the genetic libraries with SureSelect Clinical Research Exome kit by Agilent when they met quality and quantity requirements, and then they were sequenced on the Ion Proton™ System platform of Life Technologies.
The readings generated during the sequencing process were analyzed using the Torrent Suite software, where the signal produced during the sequencing process was transformed into nucleotide bases. Subsequently, a genetic mapping was performed and compared with the reference genetic sequence; variants were detected with the Torrent Variant Caller software suite, registered in ANNOVAR, and analyzed to identify those of hematological interest.
Raw data were analyzed using the following software and versions: Torrent Mapping Alignment Program v5.4.11, Torrent Variant Caller v5.4-11, Genome Analysis Toolkit v3.8-0, Picard 2.12.2-SNAPSHOT, BEDTools v2.26.0, SAMtools 1.6, and ExomeDepth 1.1.10.
The overall list of genetic variants was subjected to a prioritization process according to their probability of causality as follows:
Variants detected in the sequencing process that met the quality criteria of the software used.
Variants identified in exonic regions and splice regions.
Nonsynonymous variants, frameshift mutation, or truncating variants.
Variants with frequency < 1/2 000 in the Genome Aggregation Database (http://gnomad.broadinstitute.org/).
Variants observed in less than 5 individuals in an internal cluster sequencing database.
Common variants identified in the two individuals analyzed.
Ethical considerations
The study took into account the ethical principles for medical research involving human subjects established in the Declaration of Helsinki,25 the ethical guidelines for biomedical research established by the Council for International Organizations of Medical Sciences,26 and the scientific, technical and administrative standards for health research set forth in Resolution 8430 of 1993 of the Colombian Ministry of Health,27 after which the research was classified as minimum risk since it did not interfere with the treatment of patients
Likewise, the research was approved by the Ethics Committee of the Faculty of Medicine of the Universidad Nacional de Colombia, in accordance with Minutes No. 010-173-17 of July 27, 2017, and the Ethics Committee of the Fundación Hospital Pediátrico de la Misericordia, in accordance with Minutes No. 006 IEC-54-17 of August 15, 2017. Both participants agreed to participate in the study, and the mother signed an informed consent form authorizing her and her son's participation.
Results
The coverage data for the sequenced region of the samples taken from the patient and his relative are shown in Table 1:
Table 1 Coverage data for the sequenced region.
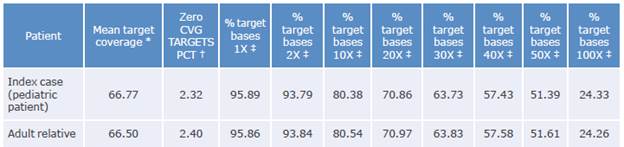
Coverage: number of times the analyzed sequence is found.
* Average readings found in sequencing.
† Fraction of unidentified reading frames.
‡ Percentage of the target bases achieving that or greater coverage.
Source: Own elaboration.
Variants present in 176 candidate genes recorded in a list created by the authors include 42 genes possibly related to SPS and platelet aggregation. The number of variants resulting from sequencing is shown in Table 2:
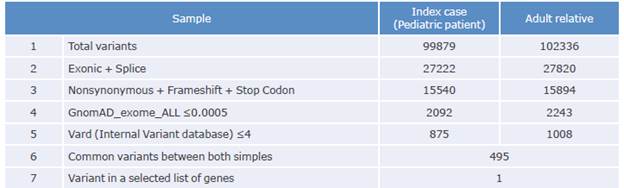
Of the total number of variants of each type found, both in the patient and in his relative, 495 were observed in both participants, and a common variant related to platelet function was identified. Table 3 shows the variant resulting from the prioritization process of the participants' samples.
Table 3 Variant resulting from the prioritization process of patient and relative samples.
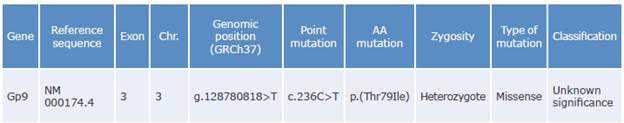
Gene: gene involved; Exon: corresponding exon number; Chr: chromosome number; Point mutation: nucleotide that changed (cytosine to thymine); AA mutation: amino acid replaced (threonine to isoleucine).
Source: Own elaboration based on Richards et al.28
The common variant identified in the index case and his relative is a cytosine to thymine transition at position c.236 (NM_000174.4) that causes the amino acid substitution from threonine to isoleucine at position p.79 of the extracellular domains of the leucine-rich repeat receptor (GPIba subunit of the glycoprotein Ib-IX-V complex), which could cause the conformational change in the main receptor of the Iba ligand, as well as platelet hyperaggregation and thrombosis. This variant has not been previously associated with SPS, although Alsahafi et al.29 had already described mutations in this gene associated with other disorders.
Figure 1 presents the report generated by the Alamut Visual software for the variant found in the participants, which provides more data on a possible association between it and SPS. Figure 2 shows the cytosine to thymine transition in the samples from both the index case and his relative.

Source: Taken from the report generated by the Almut Visual software.
Figure 1 Report of the common variant found in the participants generated by the Alamut Visual software.

Source: Document obtained during the course of the study.
Figure 2 Image of the genetic variant in the two samples analyzed using the IGV genomic viewer. Note: the image identifies the exact location of the string (at 41 bp).
These results demonstrate the coincidences of molecular tests performed on both the patient and his relative.
Discussion
Platelet adhesion, activation, and aggregation are key events in the processes of hemostasis and thrombosis, in which platelet adhesion molecules, such as the GPIb-IX-V complex, are essential.
Diseases that cause an increase in platelet function may lead to thrombotic events, such as strokes, which can kill more people than respiratory diseases and cancer.
Antiplatelet agents such as aspirin and clopidogrel, which block thromboxane and ADP receptors, are vital for the treatment of thrombosis; however, thrombotic events persist in some patients despite treatment with these drugs, making it necessary to look for other effective activation pathways. Therapies targeting other ligands may also be considered in this process.30
Brass et al.31 cite previous studies on physiological and genetic analyzes of platelet aggregation (normal and hyperaggregation states), including reports of possible genetic polymorphisms initiated by external and internal platelet stimuli, in which a relationship has been established between this process and membrane receptors of different structure, such as integrins, glycoproteins and ion channels with activation of metabolic calcium storage sequences.
The results of the present research showed a cytosine to thymine transition at position c.236 (NM_000174.4) that causes the substitution of the extracellular domains of the leucine-rich repeat receptor (GPIba subunit of the GPIb-IX-V complex) and that could also cause the conformational change of the main receptor of the Iba ligand, as well as platelet hyperaggregation and thrombosis.
It has also been described that the extracellular, transmembrane, and intracytoplasmic domains in the platelet membrane of the Ib-IX complex act as receptors responding to low thrombin concentrations and von Willebrand factor in inflammatory and infectious processes resulting in thrombus formation.32
Likewise, Gardiner33 reported that the Iba subunit is the receptor for the different ligands and acts through the N-terminal leucine-rich repeats (LRR), generating intracellular signals by itself, although binding to subunit IX promotes a fundamental conformational change to amplify these signals in thrombus formation. By the same token, Estevez et al.34 demonstrated that, by binding to thrombin, the amplification of activation in the intracellular signal pathways mediated by GPIba and GP Ibp, and GPIX is synergized by protease-activated receptors, which also act as primary receptors for thrombin and which, in inflammatory states, increase their density in the platelet membrane, increasing the possibility of thrombosis
It is important to note that intracellular mediators such as G, Racl, LIMK1 and Syk proteins, and even other tyrosine kinases enzymes such as Toll-like receptors, originated by the binding of low thrombin concentrations and the platelet von Willebrand factor to the Ib-IX-V subunit of the platelet membrane and amplified in inflammatory or infectious states, are shared by other types of platelet receptors such as ADP, thromboxane, and fibrinogen to generate thrombosis. At this point, it should be noted that the binding of thrombin to the GPIb-IX-V complex is not continuous and ends at the catalytic site of glycoprotein V, and that states of recurrent thrombosis have been observed in genetic models of absence of function of the V-ATPase subunit.35,36
It is evident that other platelet receptors should be explored given the importance of platelet activation; the relationship of platelets to inflammatory and infectious phenomena, angiogenesis, and recurrent thrombotic events despite the use of antiplatelet agents; and bleeding phenomena as side effects.37
The results obtained in the present study have not been previously reported in the literature but may be related to SPS considering the importance of glycoprotein IX in platelet functions. In this sense, it is evident that research on blockers or modulators of the GPIb-IX-V-mediated agonist response as thrombosis generators is needed.30 The main limitation of the present study is that it was carried out only in two patients diagnosed with SPS (index case and his relative); however, as a strength, this is the first time that a possible pathological variant of the disease has been described in Colombia by exome analysis.
Conclusions
In this study, it was established that both patients analyzed presented with a genetic variant consisting of a cytosine to thymine transition at position c.236 (NM_000174.4) that caused the substitution of threonine to isoleucine at position p.79 of the leucine-rich repetitive extracellular domain (GPIba subunit of the GPIb-IX-V complex). This variant has not been described previously as a finding associated with SPS, but it could be related considering the importance of glycoprotein IX in platelet functions.
In this sense, platelet hyperaggregation in SPS may not be caused by a specific mutation, but rather by the expression of several genetic polymorphisms found in patients with this disorder.
The results of the present study are highly relevant; however, further research is needed to establish the foundations of much more specific and effective treatments for the management of platelet hyperaggregation events such as SPS.

