











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
Congenital hyperinsulinism (CHI) is a term that encompasses a heterogeneous group of genetic disorders in which, due to an alteration in the regulation of insulin secretion, recurring episodes of hypoglycemia occur.1 The incidence of CHI may range from 1 case in 40 000-50 000 people in the general population to 1 case in 2 500 people in some isolated communities with high rates of consanguinity.2 One of these disorders is hyper-insulinemic/hyperammonemia (HI/HA) syndrome, which is the second most common cause of hyperinsulinemic hypoglycemia (HH) in children.3
The importance of early diagnosis and timely treatment of CHI lies in the potential for neurological damage secondary to hypoglycemic states, which may be permanent.4 Treatment involves stabilizing metabolism by administering glucose and reducing insulin secretion, which is mainly achieved using diazoxide or somatostatin analogues (octreotide). Additionally, in cases in which continuous glycemic control cannot be achieved through medical therapy, surgical treatment (pancreatectomy) should be considered.4
The present article reports the case of a newborn with hypoglycemia refractory to initial treatment and probably secondary to HI/HA syndrome. It should be noted that even though it was not possible to perform molecular characterization of the GLUD1 gene, this diagnosis was made on the basis of the patient's clinical manifestations and laboratory findings. It should also be mentioned that, in spite of the adequate response to diazoxide treatment that resulted in metabolic control, the patient presented several seizures that occurred late during hospitalization.
Case presentation
First-time mother in whom, through obstetric ultrasound, polyhydramnios and fetal macrosomia were identified. Due to the high obstetric risk, the mother was admitted to a tertiary care hospital in Bucaramanga, Colombia, where she underwent a glucose tolerance test and a A1C test, both with normal results, as well as a Doppler ultrasound to assess blood flow in the fetus and the placenta and fetal heart rate monitoring, documenting acute fetal distress. Based on these findings, it was decided to perform a cesarean section.
Once the surgical intervention was performed, a preterm male newborn was delivered (36 and 2/7 weeks of gestation) with the following characteristics: weight of 3 645gr, length of 51cm, head circumference 37cm (weight and head circumference values above the 97th and 90th percentiles, respectively, and length values between the 50-90th percentiles on the Fenton growth chart for preterm infants) (Figure 1), chest circumference of 33cm, and poor neonatal adaptation (APGAR test: 6 at one minute of birth and 8 at 5 minutes).
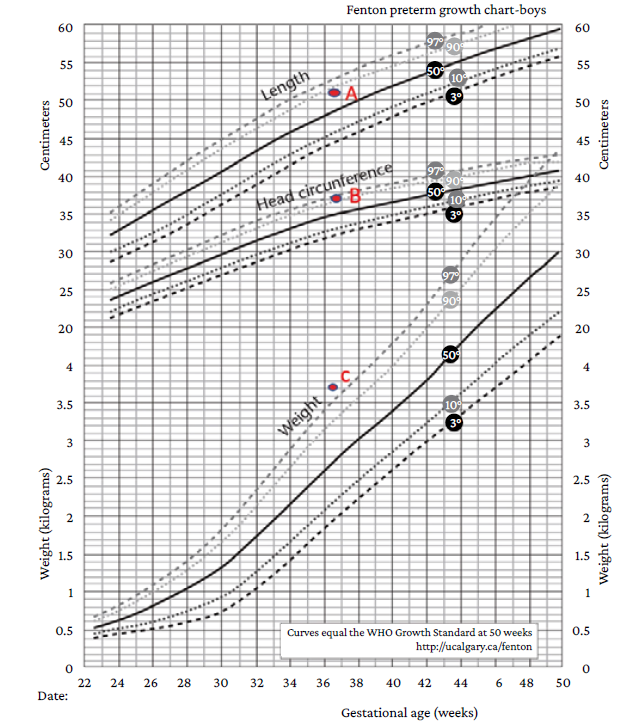
Source: Adapted from Asociación Galega de Pediatría de Atención Primaria.5
Figure 1 Fenton Preterm Growth Charts (2003). A: Length in centimeters between the 50th and 90th percentiles. B: Head circumference in centimeters above the 90th percentile. C: Weight in grams above the 97th percentile.
On physical examination, the newborn had a weak cry, hypotonia, mild respiratory distress syndrome (Silverman and Anderson test score: 3), and palpable liver 5cm below the rib cage. Upon birth, cord blood gas analysis was performed, which ruled out perinatal asphyxia, and a blood glucose test was performed at 5 minutes of life, in which a blood glucose level of 32.1 mg/dL was reported, so a bolus of 2 mL/kg of 10% dextrose was administered and a new blood glucose test was performed (97 mg/dL). One hour after bolus administration, the glucose level was 26 mg/dL, so a new bolus of 2 mL/kg of 10% dextrose was administered, followed by continuous intravenous (IV) administration of glucose (6.25 mg/kg/min).
In a new glucometry, performed 30 minutes after the start of continuous IV administration of glucose, the glucose level was 48 mg/dL, so the rate of administration was increased to 8 mg/kg/minute and IV administration of hydrocortisone (5 mg/kg/day) was started. Over the next 24 hours, despite IV administration of hydrocortisone (5 mg/kg/day), progressive increase in continuous IV administration of glucose (up to 22 mg/kg/min) and the use of total parenteral nutrition, blood sugar levels ranged from 27-70 mg/dL.
Finally, on the fifth day of life, it was possible to initiate subcutaneous administration of octreotide at 5 mcg/kg every 6 hours for a total of 20 mcg/kg/day (maximum dose), so the administration of hydrocortisone was suspended; it should be emphasized that the administration of this drug was initiated only up to this point due to the availability of the drug in the institution. Despite the treatment, there was no clinical improvement in the patient, so on the tenth day of life, several laboratory tests were performed (Table 1) using a critical sample, which yielded the following results: serum ammonia: 137.6 µmol/L; insulin: 39.1 µIU/mL; blood glucose: 26.06 mg/dL, and blood glucose/insulin ratio > 0.3, leading to confirm the diagnosis of HI/HA syndrome. Furthermore, on the same day, an echocardiogram was performed, and no abnormalities were reported. Importantly, due to administrative reasons, not all the laboratory tests requested for the critical sample were authorized.
Table 1 Laboratory tests performed on the critical sample on the tenth day of the patient's life.
| Laboratory tests | Results | Normal reference values |
|---|---|---|
| Electrolyte panel |
|
|
| Ammonia test | 137.6 µmol/L | (60-107 µmol/L) |
| Blood glucose/insulin ratio | 39.1pUI/mL / 26.06 mg/dL equal to 1.5 | (Lower than 0.3) |
| Cortisol | 193.38 ng/ml | (60-285 ng/mL in the morning) |
|
|
42 U/L 38 U/L | (6-40 U/L) (6-41 U/L) |
|
|
14 seconds 25 seconds | (10-15 seconds) (40-45 seconds) |
Source: Own elaboration.
On day 16 of life, oral administration of diazoxide (5 mg/kg/day) was started and the administration of subcutaneous octreotide was progressively decreased (1 mcg/kg every 6 hours) until reaching a dose of 3 mcg/kg every 6 hours, being completely discontinued on day 19 of life. Oral diazoxide was continued and enteral feeding was started the following day (day 20 of life), which was well tolerated by the patient, therefore, total parenteral feeding was suspended. On day 29 of life, a progressive improvement in blood glucose levels was achieved (Figure 2).
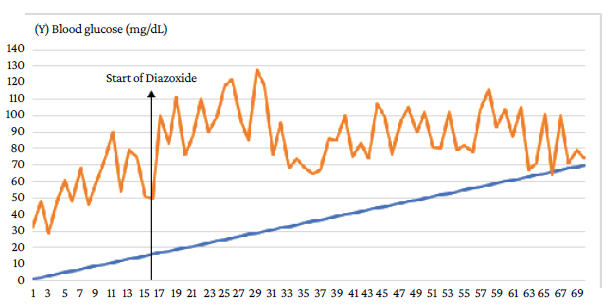
Source: Own elaboration.
Figure 2 Daily blood glucose levels of the patient during hospitalization. Response to treatment with diazoxide.
At 27 days of life, the patient had a seizure with recurrent focal episodes that later became generalized and resulted in post-seizure paralysis in the right upper limb, requiring the administration of midazolam (single dose of 0.2 mg/kg) and the initiation of maintenance doses of phenobarbital (10 mg/kg/day) and valproic acid (30 mg/kg/day), medications that led to the resolution of the seizure. Once he was stable, on the 28th day of life, a lumbar puncture was performed, which allowed to rule out neuroinfection. Then, at 30 days of life, a contrast-enhanced magnetic resonance imaging (MRI) scan of the brain was performed, and no abnormal findings were observed (Figure 3).
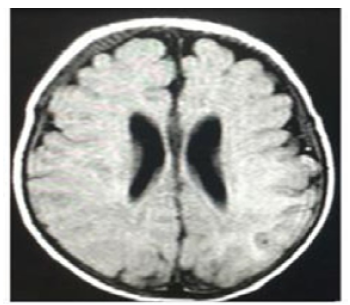
Source: Document obtained during the study.
Figure 3 Contrast-enhanced magnetic resonance imaging of the brain (gadolinium); cross-sectional view.
Later, 2 months and 5 days after birth, and since glucose levels were normal and the seizures had resolved, the patient was discharged with the following treatment regimen indefinitely: oral administration of diazoxide (10 mg/kg/day divided in two doses), phenobarbital (5 mg/kg/day divided in 2 doses), and valproic acid 30 mg/kg/day (divided in 3 doses). Additionally, an assessment by the hospital's Genetics Service and follow-up appointments with the Outpatient Service were requested; however, they were not booked since his family relocated to another city.
Discussion
Hypoglycemia is a common health issue in neonatal care units and its relevance lies in its association with neurodevelopmental disorders.6 Although there is still controversy about the cut-off point for glucose values to determine its occurrence,6 according to the Canadian Pediatric Society guidelines, hypoglycemia is defined as a glucose level <2.6 mmol/L (45 mg/dL);7 however, according to the recommendations of the Pediatric Endocrine Society,8 cognitive function is impaired when plasma glucose concentration is less than 50 mg/dL (<2.8 mmol/L).
CHI is the most common cause of persistent hypoglycemia in early childhood.9 It is a heterogeneous condition presenting with hyperinsulinism, hypoketonemia, and severe and persistent hypoglycemia.10 For its part, HI/HA syndrome, one of the heterogeneous genetic disorders encompassed by CHI,1 is a rare genetic disease caused by activating mutations in the GLUD1 gene.3 This disease is characterized by recurrent episodes of symptomatic hypoglycemia, poor fasting tolerance (less than 1 hour), a glucose/insulin ratio >0.3, and the need for IV administration of glucose at an infusion rate >8 mg/kg/minute,1,4 features that were observed in the case presented here.
Likewise, before confirming the diagnosis of HI/HA syndrome, the following differential diagnoses were considered because of their similarity to the patient's clinical manifestations: 1) hypocortisolism, which in the pediatric population is mainly caused by Addison's disease, an adrenal cortex insufficiency resulting in decreased production of glucocorticoids and mineralocorticoids,11 and 2) neonatal acute liver failure, in which clinical manifestations include growth restriction, hyporexia, hypoglycemia, coagulopathy, and cholestatic jaundice.12 However, since blood cortisol levels, liver profile results and coagulation times were normal in the present case (Table 1), these diagnoses were ruled out.
Hypoglycemia poses two problems: firstly, symptomatic treatment, as it is a medical emergency that can cause seizures, coma, irreversible neurological sequelae, or even death; secondly, the identification of the cause, as its treatment and prognosis will depend on this.13
On the other hand, it has been reported that seizures occur when glucose levels in the central nervous system drop below the normal level (80-90 mg/dL) to less than 20-30 mg/dL, a situation which, if prolonged, causes neuronal death attributable to hypoglycemia.14 However, although there is not much information on the subject, the pathophysiological mechanism of brain damage associated with HI/HA syndrome is complex and multifactorial and does not seem to be the result of a lesion directly caused by hypoglycemia.3,4,15 Possible explanations for seizure activity include persistent hypoglycemia or chronic hyperammonemia, as well as decreased levels of glutamine and the neurotransmitter GABA due to increased activity of the enzyme glutamate dehydrogenase in the brain.3,16 In this regard, it has been reported that when hypoglycemia occurs, the main excitatory amino acid glutamate is poorly reabsorbed due to its extreme secretion in the synaptic area and insufficient energy-dependent channels, resulting in increased amounts of secondary extracellular glutamate, which, in turn, induces seizures.17 In the case reported here, the absence of structural lesions on brain MRI supports this statement.
However, other studies point out that patients with HI/HA syndrome do not exhibit the central nervous system symptoms that could be expected considering their degree of hyperammonemia,18 such as drowsiness, headaches, or vomiting, which usually occur in patients with urea cycle disorders.19 One possible explanation for the absence of these symptoms in patients with HI/HA syndrome is that overactivity of glutamate dehydrogenase in the brain leads to chronic depletion of brain pools of glutamate and other amino acids that feed into the glutamate pool, especially glutamine. In this sense, if neurotoxicity due to hyperammonemia reflects osmotic shifts due to large accumulations of glutamine and glutamate, the depletion of glutamate pools in the brain could have a protective effect in these patients.20
In most cases, the onset of HI/HA syndrome occurs in the first year of life, either as a seizure secondary to hypoglycemia or as hypotonia.15 Moreover, adequate control of hypoglycemia episodes can take an average of 12 months,15 as in cases of HH, in which patients present episodes of refractory hypoglycemia, due to high insulin production, that inhibit glycolysis, gluconeogenesis, lipolysis and ketogenesis, as well as the counter-regulatory response of glucagon and cortisol by blocking KATP channels.
It has also been reported that fetal macrosomia may occur in newborns with HH due to intrauterine hyperinsulinemia and that hypertrophic cardiomyopathy and hepatomegaly may be observed in some of these patients due to increased storage of glucose as glycogen.22
In this sense, several authors highlight the need to confirm the presence of these conditions in newborns with HH, since the occurrence of hypertrophic cardiomyopathy and hepatomegaly may be related to the presence of fetal hyperinsulinemia.7,10 The patient reported in the present case had hepatomegaly, which coincides with the findings proposed by these authors.
The goal of CHI treatment is to prevent neurological damage, yet it is one of the most difficult conditions to treat.8,23 Diazoxide, in which the mechanism of action consists in opening the SUR1 subunit of the KATP channel, which reduces insulin secretion, is the pharmacological treatment of choice.24,25 Thus, early diagnosis and timely treatment is essential to prevent complications such as epilepsy, cerebral palsy, and neurodevelopmental deficits.21 In general, the treatment of IH/HA syndrome consists of a combination of a dietary regimen and pharmacological therapy (diazoxide, octreotide, or nifedipine); however, many patients are refractory to this treatment and require partial pancreatectomy.26 In the present case, the patient had a favorable response to diazoxide treatment, so surgical management was not necessary.
Although it was not possible to perform a molecular study of the GLUD1 gene in the present case, which is a limitation that should be mentioned, it could be said that, based on the clinical manifestations of the patient and the laboratory test results, it is very likely that the cause of the HI/HA syndrome was one or more mutations in this gene. Another limitation in the management of the patient was the impossibility of performing all the laboratory tests requested using the critical sample, but it is worth stressing that the diagnosis of hyperinsulinism was confirmed through the glycemia/insulin ratio and the patient's satisfactory response to the pharmacological therapy used to treat this condition.
Few studies on CHI have been carried out in Colombia, which hinders its early diagnosis and timely treatment in the country, a situation aggravated by its low prevalence. For instance, Pedraza et al.27 reported only 7 cases of CHI in a retrospective study conducted in a pediatric university hospital in Bogotá D.C. between 2012 and 2018. In view of the above, the case report presented here is particularly relevant in our country, as it provides information that will allow, in addition to identifying the clinical manifestations of patients with this form of CHI, to provide them with comprehensive care, avoiding possible complications through early diagnosis and timely treatment.
Conclusion
The HI/HA syndrome is characterized by persistent hypoglycemia, hyperinsulinism and hyperammonemia, therefore the presence of these conditions in neonates is highly suggestive of this syndrome. Its diagnosis and treatment should be timely to avoid neurological sequelae, being the transdisciplinary assessment of great importance, as it increases the chances of early diagnosis and timely administration of diazoxide (pharmacological treatment of choice) to restore normal glucose levels.

