










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Entomología
Print version ISSN 0120-0488On-line version ISSN 2665-4385
Rev. Colomb. Entomol. vol.32 no.2 Bogotá July/Dec. 2006
Caracterización molecular de líneas de Bombyx mori (Lepidoptera: Bombycidae) mediante AFLP
Molecular characterization of Bombyx mori (Lepidoptera: Bombycidae) strains by AFLP
Álvaro Alegría S.1, Enrique Aguilar F.2, Duverney Gaviria A.3, Liliana Ramírez L.4.
1 Biólogo. Asistente de investigación- CENBIOTEP-Universidad Tecnológica de Pereira E-mail:dugaar@utp.edu.co
2 Autor para correspondencia: Biólogo, Docente- Facultad Ciencias de la Salud CENBIOTEP-Universidad Tecnológica de Pereira. La Julita, Pereira-Risaralda,
Teléfono: 3215393, E-mail:eaf578@yahoo.com
3 Licenciada Biología y Química. Técnico-Centro de Desarrollo Tecnológico de la Sericultura (CDTS).
4 Doctor en Biología Molecular y Bioquímica, Director-CENBIOTEP-Universidad Tecnológica de Pereira. E-mail:AlvaroHAlegriaSA@netscape.net
Resumen. Veintitrés líneas de gusano de seda Bombyx mori L, (12 líneas de origen japonés y 11 líneas de origen chino), pertenecientes al banco de germoplasma de gusano de seda del CDTS (Centro de desarrollo tecnológico de la Sericultura) se analizaron utilizando la técnica de Polimorfismo en la Longitud de los Fragmentos Amplificados (AFLP). Se emplearon cuatro combinaciones de cebadores. Las cuatro combinaciones ensayadas, arrojaron 247 bandas con un polimorfismo promedio de 54,6%, las cuales después de ser corregidas según el criterio de Lynch y Milligan resultaron en 168 bandas con un polimorfismo de 90,4%. El dendrograma construido utilizando el algoriTmo UPGMA para los datos de disimilitud, reveló dos grupos muy bien definidos que corresponden a las razas geográficas japonesa y china. El promedio de heterocigocidad fue mayor en las líneas de la raza japonesa, observándose una dispersión mayor en el análisis de UPGMA y de los componentes principales. El estudio de la estructura poblacional, determina que aunque las dos razas no son muy diferentes entre sí, existe un componente de varianza mayor entre los grupos establecidos que dentro de éstos, lo que muestra un grado significativo de diferenciación genética.
Palabras clave: Sericultura. Estructura poblacional. Marcadores moleculares.
Abstract. Twenty-three silkworm Bombyx mori L. strains (twelve Japanese and eleven Chinese), belonging to the CDTS`s (Technological Development Center for Sericulture) silkworm germplasm bank, were analyzed using the Amplified Fragment Length Polymorphism (AFLP). Four combinations of primers were used. The assayed combinations threw 247 bands with an average polymorphism of 54.6%, these bands were corrected using Lynch and Milligan´s method, resulting in 168 bands with a polymorphism of 90,4%. The dendrogram was built using the UPGMA algorithm for the dissimilarity data, revealing two very well defined groups that correspond to the Japanese and Chinese geographical races. The heterocigocity average was higher in the Japanese race lines, showing a bigger dispersion in the UPGMA and the main components analysis. The population structure study, determines that although the two races are not very different to each other, it exists a component of greater variance, between the established groups than inside them; which shows a significant grade of genetic differentiation.
Keywords: Sericulture. Population structure. Molecular markers.
Introducción
Dentro de las diversas variedades de polillas que secretan fibras de seda, se incluyen Bombyx mori de la familia Bombycidae y las especies silvestres de la familia Saturniidae: Antherae mylitta, Antherae pernyi, Antherae assama, Antherae yamamai. Con una historia de más de 5000 años de domesticación, el gusano de seda, B mori, es la especie más importante a nivel agronómico como productor de seda; es el lepidóptero mejor estudiado debido a su rico repertorio de mutaciones bien caracterizadas que afectan virtualmente cada caracter de la morfología del organismo como también de su comportamiento y desarrollo. Su genoma haploide, con un tamaño de 560 Mb, es 3,8 veces más grande que el de Drosophila. melanogaster (Diptera: Drosophilidae) y se distribuye en 28 cromosomas holocéntricos de tamaño muy reducido (Nagaraju 2002).
El “stock” genético del gusano de seda domesticado lo componen un gran número de ecotipos y líneas autocruzadas artificialmente que se distribuyen en regiones templadas (líneas que presentan diapausa) y tropicales (líneas sin diapausa). Estas variedades difieren, entre otros, en los caracteres cualitativos y cuantitativos que afectan la calidad de la seda. Las líneas sin diapausa son pobres productoras de seda, presentan crecimiento rápido, peso corporal bajo y fibras de seda gruesas y cortas, son resistentes y se sabe que se pueden desarrollar aun en condiciones desfavorables como las encontradas en regiones tropicales, temperaturas altas y humedad alta propicias para condiciones de sanidad baja y el desarrollo de pestes y enfermedades. Las líneas tropicales de gusano de seda, sin embargo, presentan un gran número de crías al año; es decir, son polivoltinas, lo que de alguna manera compensa su productividad baja. Las líneas con diapausa, producen mayor cantidad de seda y de mejor calidad, presentan una vida larval larga, peso corporal alto y fibras de seda largas y delgadas aunque sólo se pueden obtener una o máximo dos crías al año (uni o bivoltinas). En estas ultimas, a pesar de ser mejores productoras de seda, su cría en regiones tropicales no es viable dada su alta susceptibilidad a pestes y malas condiciones de cría (Sharma et al. 1990).
Dado el trabajo que con el gusano de seda se ha llevado a cabo desde hace muchos años, en este momento se cuenta con una gran cantidad de ecotipos y más de 3.000 líneas mejoradas, y aproximadamente 400 mutantes hereditarios muchos de los cuales ya han sido mapeados (Doira 1992). La identificación y explotación de los polimorfismos al nivel de secuencias de ADN es uno de los desarrollos más significativos de la biología molecular, este tipo de polimorfismos ha sido usado con éxito en diversas especies de plantas y animales, tanto en programas de mejoramiento como identificación varietal, QTLs y mapeo genético. El uso de marcadores moleculares posee varias ventajas sobre la identificación por caracteres morfológicos, ya que no se afectan por el ambiente, y pueden ser identificados en cualquier estadio de desarrollo (Nagaraju 2000).
En el gusano de seda, se han empleado muchas técnicas para identificar el polimorfismo a nivel molecular, como el uso de marcadores enzimáticos, estudiando amilasas de hemolinfa en líneas de gusanos tropicales y de regiones templadas (Nagaraju 2000). Los análisis permitieron identificar polimorfismo electroforético de las amilasas anódicas como cuatro bandas en las líneas sin diapausa y cinco bandas en las líneas con diapausa, la banda extra en las líneas de regiones templadas corresponde a una banda de migración lenta. Cuando la separación es catódica, se identificó una sola banda en las líneas sin diapausa y ninguna en las líneas con diapausa. En este estudio también se pudo determinar que las líneas de diapausa excretan cinco veces más almidón que las líneas sin diapausa (Nagaraju 2000). Los estudios moleculares de ADN basados tanto en marcadores que usan PCR como en aquellos que no, analizando trece líneas de gusano de seda seis con diapausa y siete sin diapausa, permitieron la separación de los individuos claramente en dos ramas de un dendrograma, aquella de los individuos con diapausa y aquellos que no presentaban diapausa, al igual que permitió la identificación de marcadores exclusivos para cada uno de los dos grupos (Nagaraju et al. 1995). El análisis de hibridización con sonda Bkm-2 (GATA66TA), en las mismas trece líneas de gusano de seda, separó claramente los dos grupos y la identificación de bandas especificas para cada uno, además de la identificación de bandas específicas de sexo (Nagaraju et al. 1995). Damodar et al. (1999) informaron sobre el análisis de 28 loci microsatélites (SSR) que permitió la identificación de un marcador, sat 211, con alelos específicos para líneas con y sin diapausa, al igual que la identificación de un marcador muy polimórfico, sat 2763, con un total de 17 alelos y con una heterocigocidad elevada (90%) que permitiría fácilmente identificar las relaciones de parentesco y geográficas, muy útil, por ejemplo, en el análisis de variedades silvestres o de especies de gusano en peligro de extinción. Análisis similares sobre las mismas líneas, usando otras técnicas, han arrojado resultados parecidos (Reddy et al. 1999). Un estudio comparativo de técnicas de multilocus arrojó resultados importantes a la hora de escoger un sistema de marcadores moleculares para un trabajo en gusano de seda y aunque los agrupamientos fueron similares y se explicaban por la presencia de diapausa, origen geográfico y relaciones de parentesco de los individuos, estos agrupamientos diferían levemente en el tipo y grado de polimorfismo detectado (Nagaraju et al. 2001). El presente estudio tuvo como objetivos analizar la características poblacionales y grado de diversificación presente entre las líneas de gusano de seda B. mori mantenidas en el CDTS.
Materiales y Métodos
El estudio se desarrolló sobre 12 líneas de origen Japonés (KOI, K02, KO5, K10, K20, K30, K40, K522, SG3, SG2, NG, KNA) y 11 líneas de origen Chino (CA, CBS, CC, CJ, CGS, CHS, CLS, CTS, SC1, SC2, SC3), todas ellas pertenecientes al Centro de Desarrollo Tecnológico de la Sericultura (CDTS) mantenidas en la finca “El Pilamo”, Risaralda.
Extracción de ADN
El ADN genómico fue extraído de la porción posterior de la glándula serigena (Suzuki et al.1972). Se tomaron 0,3 g de glándula de seda de cada una de las líneas a analizar, manteniéndolas refrigeradas mientras se procesaban. El tejido se maceró en un tubo de microcentrífuga con amortiguador de extracción (NaCl 50mM, EDTA 50mM, SDS 1%) a 50°C. A cada muestra se agregaron 10 µl de proteinasa K (100 µg/ml). Las muestras se maceraron periódicamente hasta no observar fragmentos, durante la incubación en el baño. Se llevó a cabo extracción con un volumen de fenol-cloroformo-alcohol isoamilico (25-24-1) durante 5 min; posteriormente se centrifugó a 14.000 rpm durante 5 minutos y se recuperó la fase acuosa en otro tubo al que se le agregó un volumen de cloroformo-alcohol isoamilico (24-1), extrayéndose durante 5 min. Después de 5 min de centrifugación se tomó la fase acuosa y se le agregaron dos volúmenes de etanol absoluto y 1/10 de acetato de sodio 3M para precipitar los ácidos nucleicos. Posteriormente se resuspendió en amortiguador TE (10mM de Tris-HCl, 1mM de EDTA, pH 8,0) y se incubó a 37°C durante 1 h después de la adición de ARNasa (100 µg/ml). El ADN genómico fue resuspendido y cuantificado por espectrofotometría y observado por electroforesis en gel de agarosa al 0,8%.
AFLP y selección de iniciadores
Las reacciones de AFLP (Vos et al. 1995) se llevaron a cabo usando el estuche comercial ¨AFLP Analysis system I¨ No catálogo 10544-013 de la casa comercial INVITROGENE, siguiendo las recomendaciones del fabricante. La amplificación selectiva por la reacción en cadena de la polimerasa (PCR) se realizó usando una extensión de tres nucleótidos para ambos iniciadores. Las cuatro combinaciones de iniciadores empleadas, se escogieron por producir un número alto de polimorfismos no ambiguos, a partir del ensayo de 10 combinaciones sobre cuatro líneas de gusano. Los productos de PCR se mezclaron con 10 µl de solución de carga para secuenciación (98% formamida, 10mM EDTA, 0,025% xylen cyanol, 0,025% azul de bromofenol), se calentaron por 3 min a 90°C y rápidamente se enfriaron en hielo. 5 µl de la mezcla se agregaron en el gel de secuencia de poliacrilamida al 6% y se sometieron a 90 W durante 90 min. Después de terminada la corrida, el resultado se reveló mediante tinción con plata del estuche comercial “Silver sequenceTm DNA sequencing system” de la casa comercial Promega No catálogo Q-4130, siguiendo las recomendaciones del fabricante. El registro permanente de estos resultados se realizó mediante fotografía digital transfiriendo las imágenes a formato TIFF con la cámara Nikon D´100.
Análisis estadístico
Las bandas de ADN se tomaron como un locus genético asumiendo además que los alelos marcadores de diferentes loci no comigran y que cada locus puede ser tratado como un sistema de dos alelos (Lynch y Milligan 1994). De esta manera, se construyeron matrices de datos con 1 para la presencia y 0 para la ausencia de cada uno de los loci. Los datos se organizaron y analizaron por separado en razón de su origen geográfico y dentro de cada una de estas poblaciones se definieron subpoblaciones basándose en su relación de agrupamiento en el dendrograma.
Similaridad y análisis de agrupamiento
La identificación de las relaciones para las 23 líneas de gusano analizadas se basó en las matrices construidas con los índices de similaridad (Nei y Li 1979). Los valores de disimilaridad se analizaron con el método de los pares de grupos no ponderados con media ariTmética (UPGMA), y se construyó un dendrograma. Un análisis de componentes principales (Gower 1966) también se llevó a cabo para mostrar la distribución de las relaciones en tres dimensiones. Todos estos análisis se hicieron usando NTSYS-pc ver.2.01 (Rohlf 1998).
Estadística descriptiva o diversidad. Antes de correr cada uno de los análisis el supuesto de homoscedasticidad (homogeneidad de varianzas) fue revisado mediante una prueba de Bartlett (Sokal y Rohlf 1981). Los programas POPGENE, ver 1.32 (Yeh et al. 1997) y TFPGA (Tools for Populaton Genetic Analisys), ver.1.3 (Miller 1999) se usaron para calcular, bajo el supuesto de equilibrio de Hardy-Weinberg, los siguientes parámetros: (i) frecuencia del alelo recesivo (0) y consecuentemente del dominante, con el método de la expansión de Taylor (Lynch y Milligan 1994), (ii) porcentaje de loci polimórficos, (iii) promedio de heterocigocidad no sesgada de todos los loci (Nei 1978), prueba de neutralidad, prueba exacta de diferenciación poblacional (Raymond y Rousset 1995). Estos análisis se realizaron tanto con las muestras crudas, como con las cuales se les había aplicado una restricción al análisis para evitar el sesgo en bandas que presentasen una frecuencia mayor a 1-(3/N) (Lynch y Milligan 1994) datos no mostrados
Estructura poblacional. El programa AMOVA-PREP (Miller 1998) se usó para transformar el conjunto de datos dominantes para el programa de Análisis Molecular de Varianza (AMOVA, ver.1.55.; Excofier et al. 1992). Los datos se analizaron como dos poblaciones y además como dos sub-poblaciones dentro de cada una de las poblaciones, para determinar el componente aportado por cada una de las subdivisiones a los componentes de varianza total. Los análisis se realizaron tomando tanto los datos crudos como los datos corregidos con el factor 1-(3/N).
Distancia genética. La distancia genética se evaluó sobre las poblaciones y sub-poblaciones con el programa TFPGA (Tools for Population Genetic Analisys), ver.1.3 (Miller 1999), empleando el cálculo descrito por Nei (1978).
Resultados
Similaridad y análisis de agrupamiento
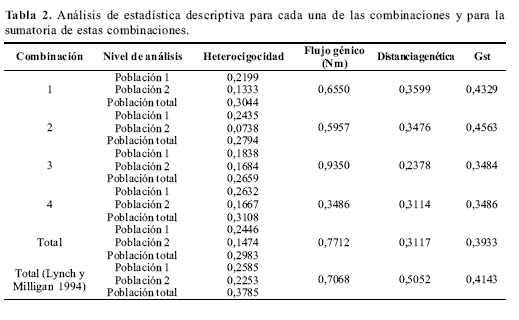
La organización de los datos se llevó a cabo definiendo subpoblaciones dentro de cada una de las razas determinado a través de la relación en el dendrograma y analizando además solamente dos poblaciones correspondientes a cada una de las razas geográficas. La significación en la escogencia de estos grupos se comprobó estadísticamente con la prueba exacta de diferenciación de poblaciones (Raymond y Rousset 1995), encontrándose que el nivel de estructuración correspondiente a subpoblaciones no era significativo, pero el nivel de población si resultó serlo. Los análisis individuales de cada una de las combinaciones usadas arrojaron resultados similares y presentaron índices de correlación entre los datos de las diferentes matrices superiores al 90%, lo que significa que el número de combinaciones empleadas es suficiente para explorar la diversidad presente en las líneas de gusano de seda. El nivel de polimorfismo para las combinaciones estuvo en el rango 65,6-75,8%, aunque se generó un gran número de fragmentos de restricción (247 bandas), a partir de las muestras individuales con la técnica de AFLP (Tabla 1). Solamente aquellos fragmentos que fueron polimórficos y se registraron sin ambigüedad se usaron para el análisis, esta corrección se hizo según el criterio establecido por Lynch y Milligan en 1994 analizándose finalmente 168 bandas de los datos totales con un promedio de polimorfismo de 90,4% (Tabla 2). El resto de los análisis se llevaron a cabo solamente para los datos corregidos.
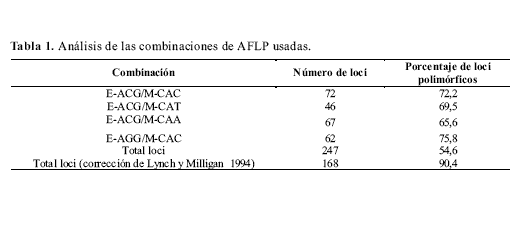
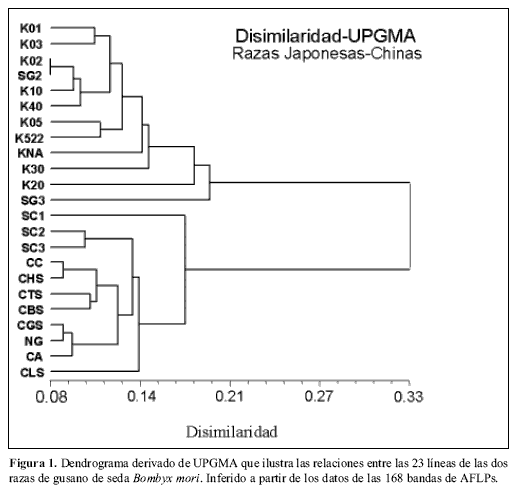
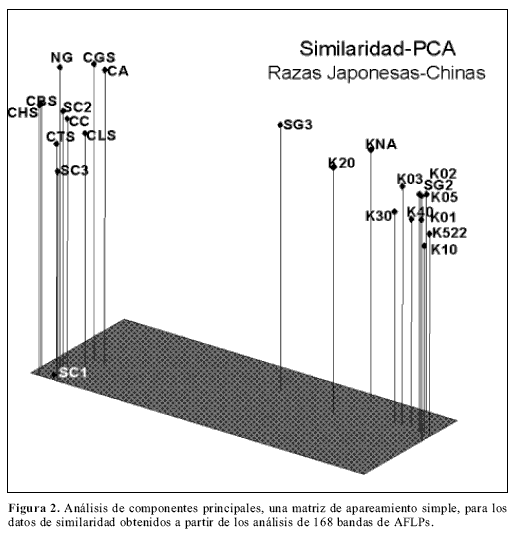
El dendrograma generado por el análisis de UPGMA de las 168 bandas para las cuatro combinaciones de iniciadores resolvió las 23 líneas de gusano de seda en dos grupos perfectamente definidos como raza japonesa y raza china (Fig. 1), a una distancia de 0,22 para las muestras de la raza japonesa y 0,18 para las muestras de la raza china. El análisis permitió identificar perfectamente a cada uno de los individuos; sin embargo, en la rama correspondiente a la raza japonesa se obtuvieron dos líneas (K02, SG2) que presentaron patrones de AFLP casi idénticos, distancia igual a 0,08 (Figs. 1 y 2).
Estadística descriptiva y diversidad
El conjunto de datos obtenido con las cuatro combinaciones de iniciadores muestra en el dendrograma y en el análisis de componentes principales, que la rama correspondiente a la raza japonesa es más heterogénea que la de la rama de la raza china. Esta observación es confirmada con los datos de heterocigocidad, los cuales siempre son mayores para las líneas de raza japonesa que para las líneas de raza china. El estimado del grado de diferenciación poblacional establecido por el valor Gst para cada uno de los loci indica un grado alto de diferenciación (Tabla 3).
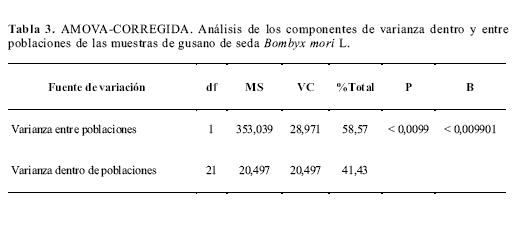
Estructura poblacional
Los componentes de varianza se obtuvieron por AMOVA (Excofier et al. 1992) a partir de las muestras corregidas, con el programa AMOVA, ver 1.55. Se analizaron las diferencias existentes entre y dentro de las dos razas evaluadas. Este análisis reveló que aunque los componente de varianza dentro y entre poblaciones son similares, el componente de varianza entre poblaciones fue un poco mayor, lo que sugiere que existe diferenciación entre las dos razas, aunque parece que el grado de diferenciación no es muy alto (Tabla 4). Este resultado se confirmó adicionalmente con una comparación de poblaciones apareadas mediante la significancia del Fst. Se realizó además el análisis de varianza para los subgrupos definidos a partir de los análisis de conglomerados y se determinó que no se comportaban como grupos, lo que significa que no existen en estas poblaciones analizadas niveles de subdivisión menor a aquellos evaluados.
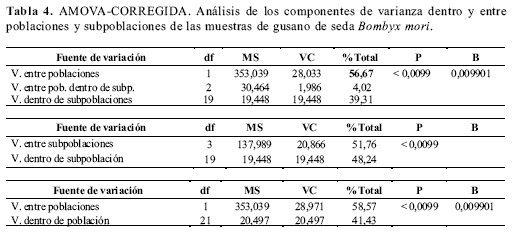
Discusión y Conclusiones
En el presente estudio se demuestra que la técnica de AFLPs puede ser usada exitosamente en gusano de seda para revelar polimorfismos a nivel del ADN útiles y que en algún momento pueden ser considerados como marcadores genéticos. Los resultados demuestran que esta técnica es valiosa en el descubrimiento de la variabilidad genética en el gusano de seda. En general, la información arrojada por cada una de las combinaciones parece ser la misma, por lo cual se puede decir que el análisis parece estar abarcando la totalidad del genoma del gusano de seda. El análisis ha permitido la identificación de bandas específicas para cada una de las razas estudiadas, las cuales pueden tener gran utilidad en establecimiento de derechos de propiedad intelectual y la determinación de pureza del germoplasma tanto en las líneas puras como en los híbridos obtenidos para uso comercial.
Los conglomerados formados identificaron perfectamente las líneas sobre la base de su origen geográfico, indicando claramente los componentes japonés y chino de cada una de las líneas, el cual parece estar acentuado debido a la estrategia de cómo son mantenidas las líneas, a través de procesos de endogamia. A pesar de que la diversidad observada fue baja, menor al 10%, los resultados permiten sugerir que las líneas a partir de las cuales se está manteniendo la raza japonesa fueron originalmente menos homogéneas que aquellas con las cuales se mantiene la raza china. Sin embargo, en el grupo de origen japonés se identificaron dos líneas casi idénticas y aunque en las líneas chinas se presentan más líneas similares entre si, no se encontró ninguna con este grado bajo de diversidad. Las variedades geográficas de gusano de seda B. mori comparten un gran componente de su genoma. Este nivel de similaridad hace necesario el estudio y evaluación de mayor cantidad de caracteres en cada una de las líneas que permita decir con mayor certeza si estas líneas, con altos niveles de similaridad, deben ser mantenidas y replicadas; es decir, si vale la pena sostenerlas como “stock” de mejoramiento. De igual manera, es importante evaluar las características de líneas como SG3, K20, K30 en la raza japonesa y sobre todo de la línea SC1 de la raza china, las cuales presentan patrones de AFLP que las ubican distantes de sus conglomerados.
Los resultados de los componentes de varianza sugieren que sí se quieren implementar programas de mejoramiento y de obtención de híbridos de mayor productividad, es necesario aumentar la base genética introduciendo nueva líneas ya sea del mismo o de diferente origen geográfico, que permitan un grado mayor de diversidad poblacional.
El gusano de seda, B. mori, comprende un gran número de ecotipos y líneas sintéticas endogámicas que muestran un alto grado de divergencia con respecto al origen geográfico, caracteres morfológicos, cuantitativos y cualitativos. Esto hace importante el estudio de la diversidad genética de varios genotipos de gusano de seda con el propósito de adelantar un programa de mejoramiento de líneas.
Agradecimientos
Los autores agradecen el apoyo financiero por parte de COLCIENCIAS del proyecto con código 1110-12-11603 y de la Universidad Tecnológica de Pereira en el marco de la convocatoria en biotecnología.
Literatura citada
ABRAHAM, E. G.; NAGARAJU, J.; DATTA, R. K. 1992. Biochemical studies of amylases in the silkworm, Bombyx mori L.: Comparative analysis in the diapausing and nondiapausing stains. Insect Biochemical and Molecular Biology 22 (8): 687-873. [ Links ]
DAMODAR, K.; ABRAHAM, E. G.; NAGARAJU, J. 1999. Microsatellite in the silkworm, Bombyx mori: Abundante, polymorphism, and strain characterization. Genome 42: 1057-1065. [ Links ]
DOIRA, H.; FUJII, H.; KAWAGUCHI, Y.; KIHARA, H.; BANNO, Y. 1992. Genetic stock and mutation of Bombyx mori. Institute of Genetic Resources, Kyushu University , Japon. [ Links ]
EXCOFIER, L.; SMOUSE, P. E.; QUATTRO, J. M. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 131: 479-491. [ Links ]
GOWER, J. C. 1966. Some distance properties of latent roots and vector methods used in multivariate analysis. Biometrika 53: 325-338 [ Links ]
LYNCH, M.; MILLIGAN, B. G. 1994. Analysis of population genetic structure with RAPD markers. Molecular Ecology 3: 91-99. [ Links ]
MILLER, M. P. 1998. AMOVA-PREP 1.01. A program for the preparation of ANOVA imput files from dominant marker raw data. Computer software distributed by the author. [ Links ]
MILLER, M. P. 1999. Tools for population genetic analysis (TFPGA) 1.3: A windows program for the analysis of allozymes and molecular population genetic data. Computer software distributed by the author. [ Links ]
NAGARAJU, J. 2000. Recent advances in molecular genetics of the silk moth, Bombyx mori. Current Science 78 (2): 151-161. [ Links ]
NAGARAJU, J. 2002. Application of the genetic principles for improving silk production. Current Science 83 (4): 409-414. [ Links ]
NAGARAJU, J.; SHARMA, A.; SETHURAMAN, B. N.; RAO, G. V.; SINGH, L. 1995. DNA fingerprinting in the silkworm Bombyx mori using banded krait minor satellite DNA-derived probe. Electrophoresis 16: 1639-1642. [ Links ]
NAGARAJU, J.; REDDY, K. D.; NAGARAJA, G. M.; SETHURAMAN B. N. 2001. Comparison of multilocus RFLPs and PCR-based markers systems for genetic analysis of the silkworm, Bombyx mori. Heredity 86: 588-597. [ Links ]
NEI, M. 1978. Estimation of average heterozygosity and genetic distance form a small number of individuals. Genetics 89: 583-590. [ Links ]
NEI, M.; LI W. H. 1979. Mathematical model for studying genetic variation in terms of the restriction endonuclease. Proc. Natl. Acad. Sci. USA. 76: 5269-5269. [ Links ]
REDDY, K. D.; NAGARAJU, J.; ABRAHAN, E. G. 1999. Genetic characterization of the silkworm Bombyx mori by simple sequence repeat (SSR)-anchored PCR. Heredity 83: 681-687. [ Links ]
ROHLF, F, J. 1998. NTSYS-PC numerical taxonomy and multivariate analysis system, Version 2.01. Owner´s manual. Exeter publication, Setauket, NY, USA. [ Links ]
RYMOND, M.; ROUSSET, F. 1995. An exact test for population differentiation. Evolution 49: 1280-1283. [ Links ]
SHARMA, A.; NIPHADKAR M. P.; NAGARAJU, J. SINGH, L. 1990. DNA fingerprint variability within and among the silkworm Bombyx mori varieties and estimation of their genetic relatedness using BKM-derived probe. The Journal of Heredity 90 (2): 315-319. [ Links ]
SOKAL, R. R.; ROHLF, F. J. 1981. Biometry. 2nd ed. Freemen, NY. 832 p. [ Links ]
SUZUKI, Y.; GAGE, L. P.; BROWN, D. D. 1972. The genes for fibroin in Bombyx mori. Journal of Molecular Biology 70: 637-649. [ Links ]
YEH, F. C.; YANG, R. C.; BOYLE, T. B.; YE, Z. H.; MAO, J. X. 1997. POPGENE, the user-friendly shareware for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton, Alta. [ Links ]
VOS, P.; HOGERS, R.; BLEEKER, M.; REIJANS, M.; VAN DEER LEE, T.; HORNES, M.; FRIJTERS, A.; POT, J.; PELEMEN, J.; KUIPER, M.; ZABEAU, M. 1995. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Research 23: 4407-4414. [ Links ]
Recibido: 30-jul-04 • Aceptado: 25-dic-05

