










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Pecuarias
Print version ISSN 0120-0690On-line version ISSN 2256-2958
Rev Colom Cienc Pecua vol.23 no.2 Medellín Apr./July 2010
Encefalopatía espongiformes transmisibles: Biología del prion y estado actual de la vigilancia epidemiológica en Colombia
Transmissible spongiform encephalopathies: biology of the Prion and current state of the epidemiologic surveillance in Colombia
Encefalopatias espongiformes transmissíveis: biologia do prião e estado atual da vigilância epidemiológica na Colômbia
Juan C Duque-Velásquez1, Biol; Andrés Villegas2, MD, PhD; Juan D Rodas1*, MV, Msc, PhD.
1Grupo Centauro, Sede de Investigaciones Universitarias, Universidad de Antioquia, Medellín, Colombia. 2Grupo de Nueurociencias, Sede de Investigaciones Universitarias, Universidad de Antioquia, Medellín, Colombia.
(Recibido: 17 julio, 2009; aceptado: 6 abril, 2010 )
Resumen
Las Encefalopatías Espongiformes Transmisibles, también llamadas enfermedades priónicas, son un grupo de enfermedades neurodegenerativas, que afectan una gran variedad de mamíferos. El agente responsable de estas patologías se ha identificado como una isoforma anormal de una proteína celular, la cual luego de sufrir un cambio conformacional (prion), adquiere la capacidad de comportarse como un agente infeccioso. Se ha demostrado la capacidad de los priones para cruzar la barrera de especies entre el ganado y los seres humanos; lo cual se ha reflejado en un problema de salud pública que ha afectado gravemente a los países donde se han presentado brotes de estas enfermedades. Las Encefalopatías Espongiformes Transmisibles se han reportado en una gran cantidad de países y Colombia no ha sido ajena a la presencia de casos esporádicos humanos, no relacionados con el consumo de material contaminado procedente de animales enfermos. Con el presente artículo se pretende dar una visión de la historia y el estado actual de Colombia frente a estas enfermedades, las cuales representan una grave amenaza para la salud pública y la agrocadena ganadera del país.
Palabras clave: Creutzfeldt-Jakob, encefalopatía espongiforme bovina, prion, scrapie.
Summary
Transmissible Spongiform Encephalopaties, also called prion diseases, are a group of neurodegenerative diseases affecting a variety of mammals. The responsible agent consists of an abnormal isoform of a cellular protein that suffers a conformational change (prion), acquiring the ability of being transmissible. It has been demonstrated prions capacity to cross the species barrier between the cattle and humans; affecting public health in countries with reported cases of bovine spongiform encephalopathy. The transmissible spongiform encephalitis have been reported in a number of countries and Colombia is not the exception with some human sporadic cases, not related with the intake of contaminated material from sick animals. With this manuscript we pretend to give a view of the history and the current state of prion diseases in Colombia, which represent a serious threat for the public health and the cattle industry of the country.
Key words: Creutzfeldt-Jakob, bovine spongiform encephalopathy, prion, scrapie.
Resumo
As doenças encefalopatias espongiformes transmissíveis, são do grupo de doenças neurodegenerativas, que afetam uma grande variedade de mamíferos. O agente responsável de estas patologias está identificado como uma isoforma anormal de uma proteína celular, a qual logo de apresentar uma mudança de conformação (prião), adquire a capacidade de comportar-se como um agente infeccioso. Tem-se demonstrado a capacidade que tem o prião para ultrapassar a barreira de espécies entre o gado e o ser humano; o qual está refletido gravemente aos países onde tem-se apresentado estas doenças. As encefalopatias espongiformes transmissíveis estão reportadas em uma grande quantidade de países. Na Colômbia tem-se apresentado casos esporádicos em humanos, não relacionados com o consumo de material contaminado procedente de animais doentes. O presente artigo, busca dar uma visão da historia e o estado atual da Colômbia frente as doenças, as quais representam uma grave ameaça para a saúde pública e da pecuária do pais.
Palavras chave: Creutzfeldt-Jakob, encefalopatia espongiforme bovina, scrapie.
¤ Para citar este artículo: Duque-Velásquez JC, Villegas A, Rodas JD. Encefalopatías espongiformes transmisibles: biología del prion y estado actual de la vigilancia epidemiológica en Colombia. Rev Colomb Cienc Pecu 2010; 240-249.
* Autor para correspondencia. Juan David Rodas. Universidad de Antioquia. Facultad de Ciencias Agrarias. Carrera 75 No. 65-87-Ciudadela de Robledo. Medellín, Colombia. Tel: (4) 2199131. E-mail address: juandavid.rodas@gmail.com.
Introducción
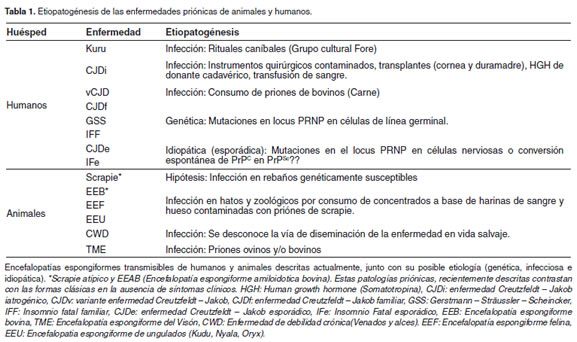
La enfermedad de Creutzfeldt-Jakob (CJD), el scrapie y la encefalopatía espongiforme bovina (EEB), son algunas de las enfermedades priónicas o encefalopatías espongiformes transmisibles (EET), un grupo desórdenes neurodegenerativos infecciosos de mamíferos, caracterizados por disfunción motora, demencia y finalmente la muerte (Weissmann et al., 2002). En animales la sintomatología consiste de cambios de comportamiento, hipersensibilidad, excitabilidad, temblores, ataxia, prurito intenso y convulsiones (Wise y Carter, 2005). El agente causal, designado prion (acrónimo: Proteinaceous Infectious particles) esta compuesto principalmente de una isoforma erróneamente plegada y parcialmente resistente a proteasas (PrPRES o PrPEET) de la proteína priónica celular (PrPC) (Prusiner, 1989). En cerebro se presenta vacuolización, astrogliósis, muerte neuronal y acumulación en el sistema nervioso central de la isoforma anormal mencionada anteriormente (Soto y Castilla, 2004). La enfermedad priónica puede desarrollarse por infección con el agente causal (vía oral, periférica y/o iatrogénica), por predisposición genética (mutaciones en el gen de la proteína priónica) e idiopáticamente (por conversión esporádica de la PrPC en PrPRES) (Prusiner, 1998) (Tabla.1). Ensayos de infección en modelos animales, han reportado infectividad en orina (Seeger et al., 2005), sangre (Hunter et al., 2002), saliva (Mathiason et al., 2006), heces fecales (Safar et al., 2008), músculo esquelético (Angers et al., 2006) y recientemente en leche (Konold et al., 2008). El estudio de estas patologías no solo tiene grandes implicaciones en el sector pecuario, económico, la salud pública, también ha contribuido al establecimiento de un nuevo principio biológico de infección, por el cual Carletón Gajdusek y Stanley Prusiner fueron galardonados con el premio Nobel de medicina en 1976 y 1997, respectivamente.
Aspectos Biológicos
Actualmente se acepta que los priones, son exclusivamente partículas protéicas infecciosas (PrPSc) desprovistas de ácidos nucléicos (Prusiner, 1997). La hipótesis de la proteína única ("proteinonly" hypothesis) esbozada por primera vez en (1976) por el matemático Ingles J. S Griffith y enunciada en su versión actualizada y detallada por Stanley B. Prusiner (1989), establece que una isoforma anormal de una proteína celular (PrPC), específica de la enfermedad (PrPSc Sc de Scrapie) constituye el principal, si no el único componente del agente infeccioso y cuenta con la capacidad de inducir la conversión de la isoforma celular PrPC en PrPSc, haciéndola insoluble en detergentes no desnaturalizantes (Meyer et al., 1986), parcialmente resistente a proteasas (Prusiner,1982) y altamente resistente a los procesos de esterilización, así como a agentes físicos y químicos capaces de degradar ácidos nucléicos virales (Alper et al., 1966, 1967).
Una de las principales evidencias a favor de la hipótesis de la proteína única es el hecho que tras la deleción del gen PrP en ratones, los individuos se desarrollan normalmente (Büeler et al.; 1992) y no sufren la enfermedad al ser inoculados experimentalmente con una cepa de scrapie adaptada en ratones (Büeler et al., 1993). La re-introducción de transgenes PrPC en la descendencia de estos ratones "Knockout", restaura la susceptibilidad a desarrollar la patología (Fischer et al.; 1996).
El gen de la proteína priónica celular humana (PRNP) se localiza en el brazo corto del cromosoma 20(20p12→pter) (Liao et al.; 1986) y codifica para una glicoproteína de membrana de un peso aproximado de 33 a 35 KDa, cuya función específica aun no esta claramente definida. Esta proteína es expresada constitutivamente en el sistema nervioso central de los mamíferos adultos (Kretzschmar et al., 1986) y en las células de los tejidos linfoides (Kitamoto et al., 1991). En infecciones por vía periférica se ha reportado la acumulación temprana de PrPSc en bazo (Clarke y Haig, 1971) y placas de peyer (Prinz et al.; 2003). Alteraciones genéticas o mecánicas (esplenectomia) de estos tejidos, incrementan el tiempo de sobrevivencia en modelos animales infectados,sugiriendo un rol importante en el proceso de neuroinvasion (Mabbott et al., 2006).
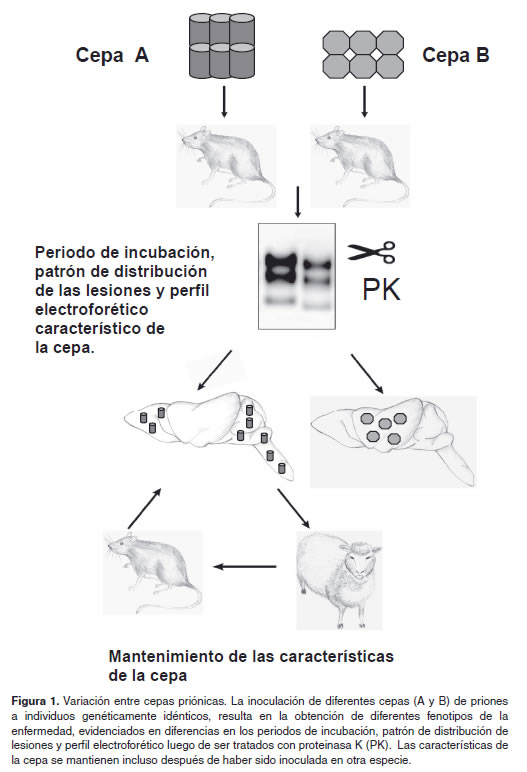
Sin embargo, existe reporte de cepas como la Fukuoka-1 capaz de realizar neuroinvasion independientemente del baso (Mohri et al., 1987). En el campo de los priones el concepto de cepa hace referencia a múltiples conformaciones de la PrPSc que al ser inoculadas de un modelo animal a otro (incluso en diferentes especies) son capaces de producir enfermedad con características especificas, tales como: Periodo de incubación, patrón de glicoformas evaluado por western blot y a nivel histológico patrón de distribución de la PrPSc, severidad y distribución de la espongiosis en cerebro (Aguzzi et al., 2007) (Figura 1). Recientemente una nueva reacción in vitro, denominada PMCA (en inglés Protein misfolding cyclic amplification), ha dado mayor fuerza a la hipótesis de la proteína única. Esta técnica utiliza una mezcla de PrPC purificada o recombinante y una semilla de PrPSc , la cual es sometida a varios ciclos de incubación y sonicación con el fin de obtener nuevas moléculas de PrPSc capaces de inducir enfermedad en modelos animales (Castilla et al., 2005).
Diagnóstico
La proteína 14-3-3 en líquido cefalorraquídeo, constituye el único marcador bioquímico incluido en los criterios de diagnostico pre-mortem de la enfermedad de CJD, reconocidos por la WHO (Golańska et al., 2008). El diagnóstico definitivo de estas enfermedades se obtiene con el análisis histopatológico y la confirmación de inmunoreactividad de la PrPSc (Budka et al., 1995). La PrPSc es el único marcador molecular fiable y su resistencia parcial a proteasas junto con la detección mediada por anticuerpos, son la base que usan las principales pruebas diagnósticas para discriminar entre la PrPC y la PrPSc (Kübler et al., 2003). Algunos de los métodos más empleados en la detección de la PrPSc son: Western Blot (Brown et al., 1994), bioensayo de transmisión de priones (Safar et al., 2002), inmunoensayo dependiente de conformación (CDI) (Safar et al., 1998), ensayo de blot celular (Bosque y Prusiner, 2000), detección inmunohistoquímica en tejidos nervioso y linfoide (Bell et al., 1997) y amplificación cíclica de plegamiento proteico erróneo (PMCA) (Saa et al., 2005). Aunque no es un signo específico, la presencia de astrocitos reactivos es considerada como marcador de apoyo en el diagnóstico definitivo (Kübler et al., 2003).
Encefalopatías espongiformes transmisibles en Colombia
En Colombia no existen datos estadísticos de la incidencia de las enfermedades priónicas, sin embargo existen reportes de un caso de Scrapie, en una oveja macho importada desde Escocia en 1981 (Citado por Toro, 2002) y en humanos un buen número de casos de CJD esporádico: El primer caso reportado en Cali, fue confirmado en el Instituto Nacional de Salud (NIH) de los Estados unidos (Zaninovic y Dueñas., 1979, 1983). A este caso se le suman tres hombres entre los 60-65 años de edad, en Cartagena, Bogotá (Toro, 1997) y Medellín (Díez et al., 1999). Colegial et al. (1999) reportan otro caso en Bogotá.
En 2006 el servicio de epidemiología del Departamento Administrativo distrital de salud de Cartagena reportó un caso confirmado en el US National Prion Disease Pathology Surveillance Center (Boletín de prensa Min. protección social No 107-06). Recientemente Villamil et al. (2007) reportaron el caso de una mujer de 65 años en Sincelejo. Adicionalmente el Instituto Nacional de Salud da reporte de varios casos de CJD esporádicos en edades entre 58 y 77 años provenientes de Cali (2005), Quindío (2005), Bogotá (2007), Medellín (2007), Betania-Antioquia (2007), Cali (2009). Dichos casos fueron evaluados en busca de la variante de la enfermedad de Creuzfeldt-jakob, como parte del sistema de vigilancia de vCJD que comparten el INS, el ICA, el Ministerio de la protección social, el INVIMA y la red nacional de bancos de sangre, sistema de vigilancia que hasta el momento sugiere la ausencia de esta patología en el país (Toro et al., 2005).
La vCJD, adaptación del agente de la EEB, hace esta patología única en el grupo de EET humanas, por acumular infectividad y PrPSc al exterior del sistema nervioso central (Ironside, 2005), y por ser la primera EET transmitida iatrogénicamente por vía sanguínea a 4 individuos quienes enfermaron luego de recibir transfusiones de sangre de individuos que fallecieron con esta patología (Llevelyn et al., 2004; Peden et al., 2004,). Las infecciones humanas suelen tener largos periodos de incubación, en ocasiones hasta de 50 años para desarrollar síntomas (Collinge et al., 2006), el portador puede diseminar la enfermedad ya sea donando sangre o sometiéndose a procedimientos quirúrgicos. En Reino Unido la epidemia de vCJD ha declinado, desde 1995 hasta 2009 existe registro de 165(4) (vivos) casos primarios y 4 (transfusión sangre) casos secundarios. A nivel mundial se han reportado dos casos en Francia, 5 en España, 4 en Irlanda, 3 en Estado Unidos y en Holanda, 2 en Portugal y 1 solo caso en países como: Italia, Canadá, Arabia Saudita y Japón (The National Creutzfeldt-Jakob Disease Surveillance Unit., 2009).
Un estudio epidemiológico reciente reporto que los componentes sanguíneos de 18 individuos, que posteriormente fallecieron de vCJD, fueron utilizados en diferentes clínicas de Inglaterra, en un total de 66 personas (44 fallecidos (4+) 22 vivos), adicionalmente se reporta que 10 casos del total de los casos de vCJD en Inglaterra, tienen historial de haber recibido transfusiones en el pasado (The Transfusión Medicine Epidemiology Review., 2009).
Programa de Vigilancia EEB en Colombia
El reporte del primer caso de EEB en 1986 en Inglaterra y su subsiguiente propagación a lo largo del Reino Unido, que posteriormente alcanzó un gran numero de países europeos, e implicó el sacrificio de aproximadamente 200.000 bovinos que padecían la enfermedad y el posterior sacrificio y destrucción de cerca de 4 millones y medio de animales asintomáticos mayores de 30 meses de edad (Brown et al.; 2001). Y su posterior transmisión a humanos, sustentan la prioridad que se le debe dar a la EEB, debido al alto impacto sanitario, social y económico que ocasionaría su diseminación.
La organización mundial de sanidad animal (OIE) y la organización mundial de la salud (OMS) han exhortado a los países a establecer medidas sanitarias para prevenir el ingreso de estas enfermedades en sus territorios y establecer un sistema de análisis de riesgos, vigilancia, y seguimiento durante 7 años. Dados los resultados del análisis de riesgo, una zona puede ser clasificada como de riesgo controlado para EEB, riesgo indeterminado y riesgo insignificante (Organización Mundial de sanidad animal, 2009).
En Colombia el Instituto Colombiano Agropecuario (ICA) estableció en 2001 el programa nacional de prevención de EEB reglamentado bajo la resolución 03153 del mismo año, la cual fija como objetivos, mantener un sistema de vigilancia epidemiológica y seguimiento continuo para la EEB, evaluar los riesgos endógenos y exógenos y establecer un sistema nacional de capacitación y divulgación sobre los aspectos concernientes a la EEB y su prevención. Establece además como obligatoria, la notificación inmediata de cualquier caso sospechoso con síntomas compatibles de esta patología, su atención inmediata y diagnóstico, la vigilancia epidemiológica se debe extender a plantas de sacrificio, predios, y empresas productoras de alimentos para rumiantes. Se debe además realizar la búsqueda, ubicación y seguimiento permanente de todos los bovinos, ovinos y caprinos importados desde países que registran o han registrado EEB (ICA, 2001).
La vigilancia pasiva de EEB comenzó en el año 1998, evaluando por histopatología 1255 casos de cuadro neurológico hasta el 2005, sin encontrar cambios morfológicos indicativos de la enfermedad. La vigilancia activa se estableció en las plantas de sacrificio en el 2002, colectando 1717 tallos encefálicos de bovinos mayores de 24 meses en 32 departamentos del país. Todas la muestras se evaluaron por histopatología, el 33%(566) se evaluó por ELISA y el 11% por inmunohistoquímicas encontrando el 100% de las muestras negativas para EEB (Sierra, 2008). Para 2003, 2004 y 2005 la OIE determinó como mínimo trabajar con 360 muestras por histopatología por año, analizándose por histopatología una total de 1127 muestras, no se realizó ELISA y se llevaron a cabo 90 inmunohistoquímicas por año, sin encontrar evidencias de la EEB (Toro et al., 2005).
Con respecto a las medidas de gestión contra riesgos endógenos, Colombia cuenta con 34 plantas registradas de producción de harinas de origen animal, dichas plantas están bajo monitoreo técnico (mínimo una vez por año) desde 2001, verificando que las condiciones de producción (temperatura, presión y tiempo de cocción) se ajusten a los estándares establecidos por la OIE y su posterior rotulado con la leyenda "prohibido su uso en la alimentación de rumiantes", cuando se trate de proteínas de origen mamífero. Adicionalmente 81 plantas de alimentos suplementarios para bovinos son objeto de vigilancia y seguimiento por parte del grupo de regulación y control de alimentos del ICA y el laboratorio nacional de insumos pecuarios (Sierra, 2008). En cuanto a riesgos exógenos, existe registro de 2918 animales que entraron al país entre 1986 y 2004 desde países que han registrado EEB en animales nativos (Francia, Alemania, Bélgica, Australia, España, Canadá). Aunque la mayoría de animales provenientes de Europa superan el tiempo de incubación de la EEB, existen importaciones recientes desde Canadá (679 animales entre 1996 y 2002) (Toro et al., 2005), de los cuales el 17% están bajo seguimiento oficial (Sierra, 2008).
Entre 1997 y 2003 han entrado al país, 3459 bovinos provenientes de Estados Unidos, país que ha reportado 2 casos en animales nativos y uno importado (Richt et al., 2007; Richt y Hall, 2008). De estos bovinos se conoce el destino de 2264 (65.4%) de los cuales 766(33.8%) se encuentran bajo seguimiento oficial. El 34.6% aun esta siendo rastreado. Suponiendo un tiempo de incubación de la enfermedad de 7 años, Sierra (2008) sugiere que los animales importados que no fueron sacrificados ya habrían desarrollado la enfermedad y deberían haber sido reportados y registrados como síndrome neurológico bovino compatible con EEB, hecho que no se reportó. Colombia esta a la espera de obtener reconocimiento de la OIE como país de riesgo insignificante (Sierra, 2008), requisito obligatorio para la exportación de ganado bovino y sus productos derivados.
Conclusiones
Gracias a su ubicación geográfica Colombia cuenta con condiciones favorables para la explotación ganadera. El numero estimado cabezas de ganado para el año 2007, fue de 23.500.000 (Fedegan. 2009), en 2005 el 57% de los animales se destinaba a la producción de carne, el 40% multipropósito y el 3% a la producción de leche (Toro et al., 2005). Un brote de encefalopatía espongiforme bovina o la aparición y confirmación de algún caso de EEB en nuestro país, no solo pondría en riesgo la salud pública de la población, aspecto bastante grave, si no que tendría graves repercusiones sobre el sector agropecuario, la industria cárnica y la economía del país.
Los esfuerzos realizados por el Instituto Colombiano Agropecuario con el fin de obtener la clasificación como país de riesgo insignificante, podrían implicar un gran avance a nivel de comercio, ya que proyectarían la industria cárnica y sus derivados, incrementando las exportaciones y permitiendo la creación de nuevos empleos. Por esta razón las acciones del programa nacional de vigilancia de EEB deben ser mantenidas y reforzadas.
Los programas de vigilancia de EEB a nivel internacional han recopilado evidencias suficientes que sugieren la presencia de dos "nuevas" cepas de EEB (BASE o EEB-L y EEB-H). A diferencia de la EEB clásica (EEB-C), estas cepas solo se han reportado en animales entre los 8 y 15 años de edad, la mayoría con signos clínicos mínimos o ausentes (Casalone, 2004). Inicialmente se sugirió que podría tratarse de cepas de baja virulencia para los humanos. Sin embargo estudios recientes en modelos animales (ratones humanizados y primates) sugieren todo lo contrario (Kong et al., 2008; Comoy et al., 2008). Todo esto aunado al prolongado periodo de incubación de la BASE con respecto a la EEB-clásica y la ausencia de síntomas clínicos evidentes, sustenta la necesidad de no interrumpir la vigilancia de los animales importados desde países con reporte de EEB en animales nativos, así el tiempo que lleven en nuestro país sobrepase el promedio de incubación reportado para la EEB clásica.
Un factor igualmente relevante y hasta hace poco desconocido, es la posible asociación encontrada entre un caso de EEB-H y una mutación en el locus PRNP (E211K), mutación también presente en la cría (Richt y Hall, 2008). Sugiriendo la posible existencia de casos de EEB de etiología genética, idea que refuerza la hipótesis de un caso genético de EEB que entro en la cadena productiva de harinas de carne y hueso propagándose a lo largo del Reino Unido en animales y humanos. Dado este reporte es imposible no preguntarse si los sistemas de vigilancia epidemiológica deberían complementarse con técnicas de genotipificación del gen de la proteína priónica bovina.
En el mismo orden de ideas, en 2006 se autorizó el ingreso de productos cárnicos y derivados procedentes de los Estados Unidos, contraviniendo las normas del ICA en el sentido de prohibir la importación de este tipo de productos desde países que hubiesen presentado casos nativos de EEB (Ministerio de industria y comercio. 2006). Cabe mencionar que un año antes, las importaciones de carne y productos relacionados provenientes de Estados Unidos se mantenían bloqueadas por sus dos principales importadores Japón y Korea (57% exportaciones de carne), tras confirmarse tres casos de EEB en dicho país en 2003, 2005 y 2006 (CRS issue brief for congress 2005).
Con el ánimo de estimular la controversia, sería importante preguntarnos que tanto se aplican las sugerencias que hace el programa nacional de vigilancia y que tanto se acatan las normas? En este sentido consideramos que un punto que debe fortalecerse en nuestro sistema de vigilancia y que es de vital importancia en el momento de reaccionar ante un brote, es la notificación de los casos sospechosos. De acuerdo con esto, valdría la pena preguntarse, que tan seguros estamos del reporte oportuno de casos, a sabiendas de que las consecuencias que un caso positivo podria acarrear serían entre otras: la inhabilitación del predio y el decomiso de los animales sospechosos o contacto. Actualmente, el sistema de compensación articulado a la declaración de emergencia sanitaria, dada la detección de un caso positivo de EEM y amparado bajo el articulo 13 de la resolución 1840 de 1994, pagará una compensación del 60% del avaluó comercial en pie de los animales, en los casos no culposos ni dolosos de emergencia sanitaria (Plan de contingencia EEB, ICA, 2008). Consideramos pues, que mientras las políticas existentes no brinden mejores garantías, la condición de país con riesgo insignificante o país libre para encefalopatías espongiformes transmisibles (EET), no pasará de ser la presunción que nos facilite el camino al comercio internacional, pero con el riego siempre implícito de fallas en el programa de vigilancia epidemiológica, pues los posibles casos no serían debidamente denunciados, si no más bien ocultados, ignorados o incinerados.
Adicionalmente, podría proponerse como una duda razonable, el preguntarse cual es la fuente de suministro actual que esta remplazando todas las harinas de carne, hueso y sangre de los bovinos y otros rumiantes, que dejaron de utilizarse como fuente de proteína en los concentrados animales, a partir de la prohibición por el riesgo de las EEB? Estamos acaso importando más harinas de pescado de los países vecinos? Estamos realmente asegurando la no utilización de los productos derivados de rumiantes como fuentes proteicas para suplementación de sus propios concentrados? Y si esto se esta haciendo, estamos también asegurando la no contaminación cruzada de estos suplementos en las líneas de producción de alimentos para rumiantes y no rumiantes de las empresas productoras de concentrados?
Como parte de los esfuerzos que se están realizando para conocer el estado actual de las enfermedades priónicas en Colombia, debemos indicar que en un esfuerzo conjunto entre los grupos Centauro y el Grupo de Neurociencias de la Universidad de Antioquia han estandarizando las técnicas de rutina para el diagnóstico de estas patologías, lo cual permitirá a los servicios de patología poder diagnosticar estas enfermedades, sin tener que acudir a laboratorios extranjeros para confirmar el diagnostico. Es de vital importancia que los servicios de patología que sospechen la presencia de un caso lo reporten al Instituto Nacional de Salud. Los grupos Centauro y Neurociencias exhortamos a los servicios médicos y de neuropatología a contactarnos en caso de encontrar casos sospechosos y sugerimos tomar muestras representativas del cerebro, cerebelo y tallo, bazo y amígdalas y almacenarlas a -70°C en viales herméticos y estos a su vez en 2 bolsas a prueba de derrames. El resto del encéfalo puede ser fijado acorde con los procedimientos de bioseguridad e histológicos de rutina.
Agradecimientos
El presente trabajo ha sido realizado gracias al aporte del Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología -Colciencias por la financiación del proyecto número: Estandarización de Técnicas Diagnósticas de enfermedades priónicas 111540820493. También queremos expresar nuestros agradeciéndoos al Instituto Nacional de Salud por los reportes facilitados.
Referencias
1. Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 2007; 8:552-61. [ Links ]
2. Angers RC, Browning SR, Seward TS, Sigurdson CJ, Miller MW, Hoover EA, Telling GC. Prions in skeletal muscles of deer with chronic wasting disease. Science 2006; 311:1117. [ Links ]
3. Alper T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun 1966; 22:278-284. [ Links ]
4. Alper T, Cramp WA, Haig DA, Clarke MC. Does the Agent of Scrapie Replicate without Nucleic Acid ? Nature 1967; 214:764-766. [ Links ]
5. Bell JE, Gentelman SM, Ironside JW, McCardle L, Lantos PL, Doey L, Lowe J, Fergusson J, Luthert P, McQuaid S, Allen IV. Prion protein immunocytochemistry – UK five centre consensus report. Neuropathol Appl Neurobiol.1997; 23:26-35. [ Links ]
6. Bosque PJ and Prusiner SB. Cultured Cell Sublines Highly Susceptible to Prion Infection. J. Virol. 2000; 74:4377- 4386. [ Links ]
7. Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima M, Bacote A, Goldfarb LG, Gajdusek DC.. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol.1994; 35:513-529. [ Links ]
8. Brown P, Will RG, Bradley R, Asher D. M, Detwiler L. Bovine Spongiform Encephalopathy and Variant Creutzfeldt-Jakob Disease: Background, Evolution, and Current Concerns. Emerging Infectious Diseases 2001; 7:6-16. [ Links ]
9. Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell - surface PrP protein. Nature 1992; 356:577- 582. [ Links ]
10. Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993; 73:1339-1347. [ Links ]
11. Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, Kretzschmar HA, Lantos PL, Masullo C, Schlote W, Tateishi J, Weller RO. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 1995; 5:459-466. [ Links ]
12. Clarke MC, Haig DA. Multiplication of scrapie agent in mouse spleen. Res Vet Sci 1971; 12:195-197. [ Links ]
13. Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Monaco S, Caramelli M. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A 2004; 101:3065-3070. [ Links ]
14. Castilla J, Saa P, Hetz C, Soto C. In Vitro Generation of Infectious Scrapie Prions. Cell 2005;121:195-206. [ Links ]
15. Colegial C, Silva F, Pérez C, Saavedra M, Fernández W, Pardo R, Lorenzana P, Vergara I. Encefalopatia por priones, caso clínico-patológico. Revista de la Facultad de Medicina de la Universidad Nacional de Colombia 1999; 4:13-20. [ Links ]
16. Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century--an acquired human prion disease with very long incubation periods. Lancet 2006; 367: 2068-2074. [ Links ]
17. Comoy EE, Casalone C, Lescoutra-Etchegaray N, Zanusso G, Freire S, Marcé D, Auvré F, Ruchoux MM, Ferrari S, Monaco S, Salès N, Caramelli M, Leboulch P, Brown P, Lasmézas CI, Deslys JP. Atypical BSE (BASE) transmitted from asymptomatic aging cattle to a primate. PLoS One 2008; 3: e3017. [ Links ]
18. CRS issue brief for congress IB10127 2005. acceso 050709 http://ncseonline.org/NLE/CRSreports/05May/IB10127.pdf. [ Links ]
19. Díez J, Jiménez A, Roselli A, Jiménez E, Ruíz N, Morales S. Enfermedad de Creutzfeldt-Jakob. Reporte de un caso. Neurociencias en Colombia 1999; 7: 49-52. Fedegan acceso 071208, www.fedegan.org.co. [ Links ]
20. Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 1996; 15:1255-1264. [ Links ]
21. Griffith JS. Self-replication and Scrapie. Nature 1967; 215:1043-1044. [ Links ]
22. Golanska E, Gresner S, Sieruta M, Liberski P. [Cerebrospinal fluid markers of prion diseases] Neurol Neurochir Pol 2008; 42:441-50. [ Links ]
23. Hunter N, Foster J, Chong A, McCutcheon S, Parnham D, Eaton S, MacKenzie C, Houston F. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 2002; 83:2897- 2905. [ Links ]
24. Ironside JW. Pathology of Variant Creutzfeldt-Jakob disease, 1-14. Prions: Food and Drug safety. Kitamoto (Ed) Springer-Verlag Tokyo 2005. [ Links ]
25. ICA -Instituto Colombiano Agropecuario. Informe de gerencia año 2001. 2002 acces Julio 5 de 2009. URL: http://www.ica.gov.co/Gestion/Infogerencia /INFORMEGERCIA01.PDF [ Links ]
26. Kitamoto T, Muramoto T, Mohri S, Dohura K, Tateishi J. Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. J. Virol 1991; 65:6292–6295. [ Links ]
27. Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ.Scrapie prion proteins are synthesized in neurons. American. J. Pathol 1986; 122:1-5. [ Links ]
28. Kübler E, Oesch B, Raeber AJ. Diagnosis of prion diseases. Br Med Bull. 2003; 66:267-279. [ Links ]
29. Konold T, Moore SJ, Bellworthy SJ, Simmons HA. Evidence of scrapie transmission via milk. BMC Vet Res. 2008; 4:14. [ Links ]
30. Kong Q, Zheng M, Casalone C, Qing L, Huang S, Chakraborty B, Wang P, Chen F, Cali I, Corona C, Martucci F, Iulini B, Acutis P, Wang L, Liang J, Wang M, Li X, Monaco S, Zanusso G, Zou WQ, Caramelli M, Gambetti P. Evaluation of the human transmission risk of an atypical bovine spongiform encephalopathy prion strain. J Virol 2008; 82:3697-3701. [ Links ]
31. Liao J, Yu-Cheng, Lero VR, Clawson GA, Smuckler EA. Human Prion protein cDNA: Molecular Cloning, Chromosomal Mapping, and Biological Implications. Science 1986; 233:364-367. [ Links ]
32. Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004; 363:417-421. [ Links ]
33. Mabbott NA, MacPherson GG. Prions and their lethal journey to the brain. Nat Rev Microbiol 2006; 4: 201-211. [ Links ]
34. Mathiason CK, Powers JG, Dahmes SJ, Osborn DA, Miller KV, Warren RJ, Mason GL, Hays SA, Hayes-Klug J, Seelig DM, Wild MA, Wolfe LL, Spraker TR, Miller MW, Sigurdson CJ, Telling GC, Hoover EA. Infectious Prions in the Saliva and Blood of Deer with Chronic Wasting Disease. Science. 2006; 314:133-136. [ Links ]
35. Meyer RK, Mckinley MP, Bowman KA, Braunfeld MB, Barry RA, Prusiner SB. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986; 83:2310-2314. Ministerio de protección social Boletín de prensa No 107-06 Acceso: Julio 10 de 2009 http://www.minproteccionsocial. gov.co/VBeContent/NewsDetail.asp?ID=15245&IDCompany=3 Boletin de prensa No 107-06. [ Links ]
36. Ministerio de Indrustria y comercio republica de Colombia. Acceso 0702 http://www.mincomercio.gov.co/econtent/Documentos/negociaciones/TLC/Care/CartaFinalSigned.pdf. [ Links ]
37. Mohri S, Handa S, Tateishi J. Lack of effect of thymus and spleen on the incubation period of Creutzfeldt-Jakob disease in mice. J Gen Virol 1987; 68: 1187-1189. [ Links ]
38. Organización Mundial de sanidad animal. Accseso 070909. http://www.oie.int/eng/maladies/en_alpha.htm?e1d7. Plan de contingencia-Encefalopatia epsongiforme Bovina. ICA. 2008. [ Links ]
39. Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364:527-529. [ Links ]
40. Prinz M, Huber G, Macpherson AJ, Heppner FL, Glatzel M, Eugster HP, Wagner N, Aguzzi A. Oral prion infection requires normal numbers of Peyers patches but not of enteric lymphocytes. Am J Pathol 2003; 162:1103-1111. [ Links ]
41. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982; 216:136-144. [ Links ]
42. Prusiner SB. Scrapie Prions. Annu. Rev. Microbiol. 1989;43:345-374. [ Links ]
43. Prusiner SB. Prions. Nobel Lecture. Proc. Natl. Acad. Sci. USA. 1998; 95:13363-13383. [ Links ]
44. Prusiner SB. Prion Diseases and the BSE Crisis. Science. 1997; 278:245-251. [ Links ]
45. Richt JA, Kunkle RA, Alt D, Nicholson EM, Hamir AN, Czub S, Kluge J, Davis AJ, Hall SM. Identification and characterization of two bovine spongiform encephalopathy cases diagnosed in the United States. J Vet Diagn Invest 2007; 19:142-54. [ Links ]
46. Richt JA, Hall SM. BSE case associated with prion protein gene mutation. PLoS Pathog 2008; 4: e1000156. [ Links ]
47. Saa P, Castilla J, Soto C. Cyclic amplification of protein misfolding and aggregation. Methods. Mol. Biol. 2005; 299:53-65. [ Links ]
48. Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrPSc molecules with different conformations. Nature. Med. 1998; 4:1157-1165. [ Links ]
49. Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, Serban H, Groth D, Burton DR, Prusiner SB, Williamson RA. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nature Biotechnol 2002; 20:1147-1150. [ Links ]
50. Safar JG, Lessard P, Tamgüney G, Freyman Y, Deering C, Letessier F, Dearmond SJ, Prusiner SB. Transmission and detection of prions in feces. J Infect Dis.2008; 198: 81-89. [ Links ]
51. Seeger H, Heikenwalder M, Zeller N, Kran ich J, Schwarz P, Gaspert A, Seifert B, Miele G, Aguzzi A. Coincident Scrapie Infection and Nephritis Lead to Urinary Prion Excretion. Science. 2005; 310:324-326. [ Links ]
52. Sierra. U. E. Avance y estado actual de la prevencion y vigilancia de la encefalopatia espongiforme bovina (EEB) en Colombia. En Jornada nacional de zoonosis y enfermedades emergentes y re-emergentes. 2008. Acceso Julio 14 de 2009. http://sites.google.com/site/saludpublicaveterinaria/. [ Links ]
53. Soto C, Castilla J. The Controversial Protein-Only Hypothesis of Prion Popagation. Suplement. Nature medicine. 2004; 10: 563-567. The National Creutzfeldt-Jakob Disease Surveillance Unit. Accseso 071109. http://www.cjd.ed.ac.uk/vcjdworld.htm. [ Links ]
54. The Transfusion Medicine Epidemiology Review. Acceso 071109. http://www.cjd.ed.ac.uk/TMER/TMER.htm [ Links ]
55. Toro G. Demencia – Priónes y Enfermedades Priónicas. Referencia especial a las vacas locas. Revista de la academia Colombiana de Ciencias Exactas Físicas y Naturales 1997; 21:229-236. [ Links ]
56. Toro G, Díaz A, Saad C. Ayer, Hoy y Mañana, la Teoría del Prión. Acta Neurológica Colombiana 2002; 18:187. [ Links ]
57. Toro G, Pacheco OE, Sierra UE, Beltrán M, Díaz A, Parra EA, Bonilla E. Encefalopatías subagudas espongiformes transmisibles (ESET). La Teoría Prión -Enfermedades priónicas. Acta Neurol Colomb. 2005; 21:135-162. [ Links ]
58. Villamil W, González J, Arrieta JA, Álvarez C, Borja G, Vergara JC, Buelvas D. Enfermedad de Creutzfeldt- Jakob tipo esporádica: reporte de caso. Infectio 2007; 11:124-128. [ Links ]
59. Weissman C, Enari M, Klöhn PC, Flechsig E. Transmission of Prions. Proc. Natl. Acad. Sci. USA. 2002; 99:16378-16383. [ Links ]
60. Wise DJ y Carter GR. Prions and transmissible spongiform encephalopaties. International Veterinary Information Service, In: A Concise Review of Veterinary Virology (www.ivis.org). 2005. [ Links ]
61. Zaninovic V, Dueñas A. Un caso de Enfermedad de Creutzfeldt-Jakob. Acta Médica del valle 1979; 10: 131-36. [ Links ]
62. Zaninovic V, Dueñas A. Confirmación de un caso de Enfermedad de Creutzfeldt-Jakob. Colombia Medica1983; 14:40-2. [ Links ]

