










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Ciencias Pecuarias
Print version ISSN 0120-0690On-line version ISSN 2256-2958
Rev Colom Cienc Pecua vol.24 no.4 Medellín Oct./Dec. 2011
Detection of seven viruses and Mycoplasma in fetal bovine serum by real time PCR¤
Detección de siete virus y de Mycoplasma en suero fetal bovino por PCR en tiempo real
Detecção de sete vírus e de Mycoplasma em soro fetal bovino por PCR em tempo real.
Claudia P Cordero Camacho1, QF, DrSc; Linamaría Escobar Mármol1, Biol, MSc (C); Edward F Carrillo Borda1, Biol, DrSc; Sandra J Morantes Medina1, Biol, DrSc (C); Fabio A Aristizábal Gutiérrez1*, QF, DrSc.
1 Grupo de Farmacogenética del Cáncer, Departamento de Farmacia, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá.
(Recibido: 2 febrero,2011; aceptado: 23 mayo, 2011)
Summary
Objective: real time PCR analysis for the detection of seven bovine pathogenic viruses: Bovine Adenovirus (BAdV), Bovine Viral Diarrhea Virus Types 1 and 2 (BVDV-1 and BVDV-2), Bovine Respiratory Syncytial Virus (BRSV), Vesicular Stomatitis Virus (VSV), Bovine Parainfluenza Virus 3 (BPIV-3), Bovine Herpes Virus-1 (BoHV-1), and Mycoplasma was conducted using fetal bovine serum (FBS, MICROGEN®) obtained in Colombia, aiming to include it as part of the serum quality control. Methods: bovine derived MDBK and human derived HEp-2 cell lines were cultured with the test serum for 21 days, collecting supernatant and cellular samples every 7-days. Once DNA and RNA were extracted, the later was converted into cDNA and both samples were subjected to real time PCR using specific primers and Resolight® (DNA-binding fluorescent dye). Standard curves were generated using serial dilutions of cloned specific viral sequences. Accurate amplification and high efficiency was demonstrated in these reactions. Results: realtime PCR amplification did not show a persistent increase of viral counts in cultures during the 21-day follow-up. However, for vesicular stomatitis virus, a transient increase was observed at 7 and 14 days in both cell lines, but considered as not conclusive for viral presence. Conclusions: real time PCR analysis showed to be a suitable method for viral detection in fetal bovine serum samples and through this method no consistent viral or mycoplasma presence was detected in the MICROGEN® fetal bovine serum.
Key words: bovine virus, fetal bovine serum, real time PCR, viral detection.
Resumen
Objetivos: se empleó el método de PCR en tiempo real para detectar los virus patógenos bovinos: Adenovirus bovino (BAdV), virus de la diarrea Viral Bovina tipos 1 y 2 (BVDV-1 y BVDV-2), Virus Respiratorio Sincitial Bovino (BRSV), Virus de la Estomatitis Vesicular (VSV), Virus de la Parainfluenza Bovina tipo 3 (BPIV-3), Herpesvirus Bovino-1 (BoHV-1) y Mycoplasma, en suero fetal bovino (FBS, MICROGEN ®)obtenido en Colombia, con el objetivo de incluir estos análisis en el control de calidad del FBS. Metodos: las líneas celulares MDBK de origen bovino y HEp-2 de origen humano se cultivaron con el FBS MICROGEN® por 21 días, tomando muestras de cultivos y sobrenadantes cada 7 días. Una ves extraido el DNA y RNA, a partir de este último se sintetizó cDNA, y en los dos tipos de muestras se analizó la presencia de los agentes patógenos mencionados por PCR en tiempo real empleando iniciadores específicos para cada uno y Resolight® (colorante fluorescente de unión a DNA). Se generaron curvas estándar con diluciones seriadas de secuencias virales específicas clonadas en plásmidos, que mostraron amplificación específica y altas eficiencias. Resultados: el análisis de los cultivos mantenidos con el FBS en estudio no mostró aumento del número de copias virales detectadas a lo largo del periodo de 21 días de seguimiento, excepto para el virus de la estomatitis vesicular, que mostró un incremento transitorio en los sobrenadantes de los cultivos de las dos líneas celulares a los 7 y 14 días de cultivo, que no se consideró concluyente para la presencia del virus. Conclusiones: el método de PCR en tiempo real mostró utilidad para la detección de virus patógenos y mycoplasma en FBS, y mediante este método no se obtuvieron resultados que permitan concluir que los patógenos virales o los mycoplasmas están presentes en los cultivos mantenidos con el suero fetal bovino MICROGEN®.
Palabras clave: detección viral, PCR en tiempo real, suero fetal bovino, virus bovinos.
Resumo
Objetivo: Foi utilizado o método de PCR em tempo real para detectar os vírus patogénicos bovinos: Adenovírus Bovino (BAdV), Vírus da Diarreia Viral (BVDV-1 e BVDV-2), Vírus Respiratório Sincicial Bovino (BRSV ), Vírus da Estomatite Vesicular (VSV), Vírus da Parainfluenza Bovina tipo 3 (BPIV- 3), Herpesvírus Bovino-1 (BoHV-1) e Mycoplasma, em soro fetal bovino (FBS, Microgen®) obtido na Colômbia, de modo a incluir esta análise no controle de qualidade FBS. Métodos: as linhas celulares de MDBK de origem bovina e HEp-2 de origem humana foram cultivadas com FBS Microgen® durante 21 dias, tomando amostras de cultura e sobrenadantes cada 7 dias. Uma vez retirado o DNA e RNA, foi sintetizado o cDNA a partir do RNA. Nos dois tipos de amostras foram analisadas para determinar a presença de patógenos mencionados por PCR em tempo real usando primers específicos para cada um e Resolight®(corante fluorescente de união à DNA). As curvas padrão foram geradas com diluições em série de sequências virais específicas, clonadas em Resultados: plasmídeos, que mostram amplificação específica e altas eficiências. a análise das culturas mantidas em FBS em estudo, não mostraram aumento no número de cópias virais detectadas ao longo do período de 21 dias de seguimento, exceto para o vírus da estomatite vesicular, que mostrou um aumento transitório nos sobrenadantes das culturas de duas linhas celulares aos 7 e 14 dias de cultura, que não foi considerado conclusivo para a presença do vírus. Conclusões: O método de PCR em tempo real foi útil para a detecção de vírus patogênicos e mycoplasma em FBS, e por este método foram obtidos resultados que demonstram que patógenos virais ou mycoplasmas estão presentes nas culturas mantidas com soro fetal bovino Microgen®.
Palavras chave: soro fetal bovino, vírus bovinos.
¤ To cite this article: Cordero CP, Escobar L, Carrillo EF, Morantes SJ, Aristizábal FA. Detection of seven viruses and Mycoplasma in fetal bovine serum by real time PCR. Rev Colomb Cienc Pecu 2011; 24:585-597
* Corresponding author: Fabio Aristizábal Gutiérrez. Departamento de Farmacia, Universidad Nacional de Colombia, sede Bogotá. Ciudad Universitaria. Cra 30 # 45-03.Edificio 450. Oficina 206. Bogotá D.C. Colombia, Suramérica. E-mail:cpcorderoc@unal.edu.co, faaristizabalg@bt.unal.edu.co
Introduction
Fetal bovine serum (FBS) is an essential supplement for in vitro cell culture medium, as a source of growth factors, vitamins, minerals and hormones that stimulate cell proliferation. Major concerns on FBS are related with high cost and presence of bovine viruses and Mycoplasma, non desired agents that may alter normal course of cell cultures. An alternative to this potential problem has been the use of serum-free media, however, associated high costs are a major drawback (Freshney, 2000).
In Colombia the FBS requirements have been satisfied by international suppliers as GIBCO-BRL-Invitrogen®, SIGMA®, Eurobio®, Lonza and Hyclone®, which accounts for a high price of the product and increases culture maintenance expenses. As an alternative to foreign produced and imported fetal bovine serum the Colombian company MICROGEN® produces a Colombian fetal bovine serum and offers this product to the internal market and the national scientific community.
In addition to sterility and basic biochemical quality controls, MICROGEN® FBS should be evaluated for culture performance and virus absence, in order to ascertain its suitability for use in cell cultures.
Viral detection in bovine serum was based on antigen detection through ELISA and other immunological methods; within the last 15 years, RNA and DNA detection methods by real time RT- PCR have been proved to be more sensitive as well as highly specific (APHIS-USDA, 1995; Kosinova et al., 2007; Timsit et al., 2010).
The present work was intended to establish a real time RT-PCR analytical method for the detection of bovine viruses and mycoplasma and its application in the analysis of commercial batches of the MICROGEN® FBS.
Materials and methods
Virus selection
Bovine viruses to be detected in the MICROGEN® FBS were selected based on the available Colombian epidemiological information on bovine viral infections showing high prevalence (Betancur et al., 2010, Cajas, et al., 2003, Piedrahita et al., 2010, Vargas et al., 2009) and on the list of viruses analyzed by GIBCO FBS manufacturer (GIBCO, 2007). The seven viruses included were BAdV, BVDV-1 and BVDV-2, BRSV, VSV, BPIV- 3, BoHV-1.
Mycoplasma were included as those intracellular parasites are potential contaminants of animal derived products and some of them can be present in cattle's blood (Freshney, 2000; Nishizawa, 2010).
Selection of primers for real time PCR
Specific primers for real time PCR-detection were selected based on previous reports of real time analysis for the target viruses and basic primer- design requirements: selection of conserved regions as target sequences, melting temperature (Tm) close to 60 °C and PCR product length between 100 and 250 bp. Primer3 software (Rozen and Skaletsky, 2000) and the PrimerQuest tool included in the IDT- SciTools (IDT, 2009), were used for primers design and selection. Primers were synthesized by IDT (IDT, Coralville, IA).
Construction of positive controls
Two oligonucleotides containing the target sequences of the primers were designed, synthetized and ligated to suitable vectors. Oligonucleotide A (OligoA) containing target sequences for VSV, BoHV-1, BVDV-I and BPIV-3 primers, was cloned in the pGEM®-T Easy cloning vector (Promega, Madison, WI). Oligonucleotide B (OligoB), containing target sequences for BAdV, BRSV, BVDV-2 and mycoplasma primers was cloned in the pCR® 2.1- TOPO® cloning vector (Invitrogen, Carlsbad,CA). Cloning procedure was conducted following manufacturer's instructions. Both recombinant constructs were transformed in DH-5α Escherichia coli competent cells, purified with the Mini-prep Wizard (Promega, Madison, WI) and quantified by Qubit®(Invitrogen, Carlsbad,CA).
Generation of standard curves
A standard curve was constructed for each one of the studied bovine viruses and mycoplasma using eight serial 10-fold dilutions of cloned OligoA or OligoB. HRM Master, containing the DNA binding dye ResoLight® (Roche, Mannheim, Germany) was used for real time detection with excitation wavelength 465 nm and emission wavelength 510 nm. Reactions were carried out in 10 µL final volume, in the LightCycler 480 Instrument II using multiwell 96 plates® (Roche, Mannheim, Germany).
Real time PCR reactions included 1x HRM Master Mix, 2.5 mM MgCl2, 0.2 µM each specific primer, 2.0 µL of quantified cloned control oligonucleotide and ultra pure PCR-grade water to a final volume of 10 µL. Each reaction was carried out in triplicate. The real time PCR amplification program included: pre-incubation 95 °C for 10 min, 45 amplification cycles: 95°C for 10 s, 60°C for 15 s, 72°C for 10 s, followed by one melting cycle: 95°C for 1 min, 40 °C for 1 min, 60 °C for 1 s, 95°C and cooling to 40 °C.
Amplification curves, Ct values, slope values of the curves and derivative melting curves were obtained with the LightCycler 480® II data analysis software (Roche, Mannheim, Germany). Efficiency percentage for each standard curve was determined by the equation E= (10 (-1/m) – 1) x 100, where m is the slope value of the curve. Ct value for each reaction, the amplification cycle at which fluorescence crosses the threshold line, was used as the measurement parameter and plotted against plasmid copy number in the Microsoft Office Excel application (Microsoft, Redmond, WA).
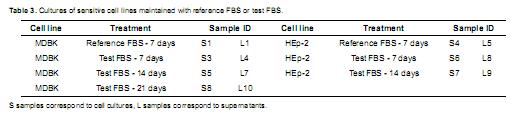
Preparation of samples Cell culture.
Cultures of cell lines sensitive to infection by the studied viruses were maintained with reference or test fetal bovine serum. MDBK (bovine kidney derived), and HEp-2 (human larynx cancer derived) cell lines (ATCC, Manassas, VA) were maintained in Eagle's MEM medium (SIGMA, St Louis, MO), supplemented with penicillin100 U.mL-1, streptomycin 100 µg. mL-1 (Gibco-BRL , Carlsbad,CA) and 10% reference fetal bovine serum Certified-FBS (Gibco-BRL®, Carlsbad,CA) or test FBS MICROGEN® (Microgen, Bogotá, Colombia). Cultures were maintained in 75 cm2 culture flasks (TPP, Trasadingen, Switzerland) in standard culture conditions 37 °C, 5 % CO2 in air, 100% humidity. Media was changed once a week.
Cell cultures were maintained with reference FBS for 7 days or with test FBS MICROGEN® for 14 to 21 days. Cellular and supernatant samples were taken from each culture when subcultured at 7, 14 and 21 days of maintenance and processed for total RNA and DNA extraction.
This cell culture procedure was based on the procedure used for viral testing by Gibco-BRL® accoding to the Requirements for ingredients of animal origin (APHIS-USDA,1995).
RNA and DNA extraction. Total RNA and DNA were extracted from culture samples using the phenol-guanidine isothiocyanate-chloroform method. 1 x 105 cells (solid samples) were mixed with 1 mL of TRIzol® reagent (Invitrogen, Carlsbad, CA) and 1 mL of supernatant (liquid samples) was mixed with 3 mL of TRIzol® LS reagent (Invitrogen, Carlsbad, CA). Extraction was made according to the manufacturer's instructions. Total RNA was resuspended in 20 µL of nuclease-free DEPC-treated water; DNA was resuspended in 8 mM NaOH solution added with HEPES and EDTA solutions (Invitrogen, Carlsbad, CA). RNA and DNA samples were quantified with the Qubit fluorometric method (Invitrogen, Carlsbad, CA).
Reverse transcription. RNA was converted into cDNA using the SuperScript III First-strand system (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. Each RT reaction included: 1X RT buffer, 5 mM DTT, RNAse inhibitor 40 U, dNTPs 0.5 mM each, random hexamers 50 ng, SuperScript III reverse transcriptase 200 U, 2.5 µg total RNA and DEPC- treated water for a final volume of 20 µL. Reactions were incubated at 25 °C for 5 min, 50 °C for 60 min and 70 °C for 15 min. RT products were diluted 1:2 with DEPC-treated water. A conventional PCR with -actin specific primers was made to verify cDNA synthesis, using the RT reaction product as template.
Similar PCR reactions were made using genomic DNA as template to verify the suitability of these samples as PCR substrates. Each conventional PCR reaction contained 1X Taq polymerase buffer, 2mM MgCl2, 0.2 mM each dNTPs, 0.1 µM forward primer (GGCACCCAGCACAATGAA GATCAA), 0.1 µM reverse primer (ACTCGTCATACTCC TGCTTGCTGA), 1µL RT reaction product or 100 ng genomic DNA, 0.5 U Go-Taq polymerase (Promega, Madison, WI), and PCR-grade water to 25 µL final volume. Amplification reactions were performed in the My Cycler® thermal cycler (BIORAD, Hercules, CA); amplification program started with a denaturation step of 94 °C for 2 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 15 s, 72 °C for 10 s. PCR products were evaluated by electrophoresis in 2% agarose gels, stained with SYBR Safe® (Invitrogen, Carlsbad, CA) and visualized in a Digimage® (Major Science, Saratoga, CA) gel documentation system. The expected sizes for the PCR products were 121 bp for bovine derived cells and 133 bp for human derived cells.
Real time PCR Assay
Real time PCR detection of the seven viruses and mycoplasma were performed using cDNA as template, BAdV, BoHV-1 and Mycoplasma detection was made also in DNA. Real time reactions were set up using the same conditions described for the construction of standard curves, using 2 µL of diluted cDNA product as template or 10 ng of total DNA. In negative controls nuclease- free water was added as template instead of sample. In positive controls dilutions of the correspondent control oligo were added as template. Each sample was evaluated in duplicate. For each reaction the Ct value was determined and Tm for the product was also observed.
To avoid cross contamination all technical procedures were held under controlled conditions, under laminar flow hoods and carried out in laboratories where no other viruses were under investigation.
Statistical analysis
Linear regression for each standard curve was obtained and the concentration of each sample was automatically calculated based on linear equation, using the LightCycler 480® II data analysis software (Roche, Mannheim, Germany).
Results
Selection of primers for real time PCR
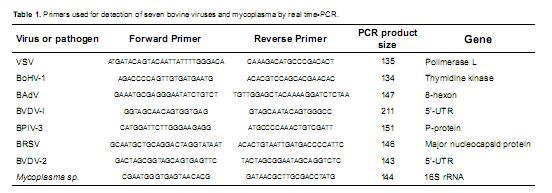
The sequences of the specific primers selected for the real time PCR reactions are presented in table 1.
Standard curves
Standard curves were obtained using each specific primers pair (Table 1) and the correspondent control oligonucleotide A or B (Figures 1 and 2).
As serial dilutions of the cloned control oligonucleotides were used for standard curves' construction, the amount of template was expressed as copies number of the recombinant plasmid containing the target sequence: ranging from 2 to 2 x 107 target copies. Amplification curves for each set of serial dilutions showed that increasing Ct values were obtained as the copy number in the dilution decreased (Figure 1). A linear trend was observed for each dilution series when Ct values were plotted against the copy number (Figures 2 and 3). Melting curves obtained for the products of these reactions showed a unique peak indicating the absence of primer-dimmers and non-specific amplification (Melting curves not shown).
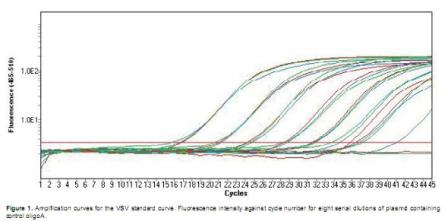
In the standard curves obtained with control oligoA corresponding to VSV, BVDV-1, BoHV-1 and BPIV-3, the linear pattern was lost, or no amplification was detected, for the lower dilution (two copies), therefore 20 copies dilution was considered as the lower detection limit for these three viruses (Figure 2).
Small standard deviation values were obtained in the four data sets and regression coefficient (R2) wasabove 0.98, indicating high adjustment to the linear model.
In the standard curves constructed with oligoB the linear pattern was conserved for the 20 copies dilution but not for two copies dilution, which showed lower Ct values than the ones obtained for the 20 copies dilutions.
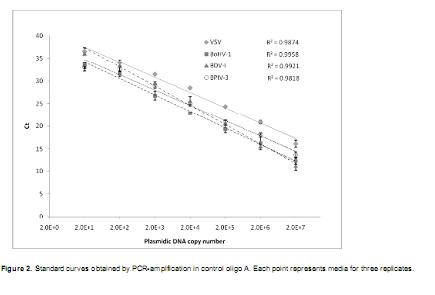
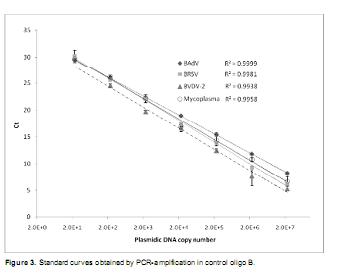
Based on these results the detection limit for BAdV, BRSV and mycoplasma was established at 20 copies. For BVDV-2 no amplification was detected for 2 and 20 dilutions thus the detection limit was set at 200 copies. Ct values decreased proportionally to copy number increase, including the highest concentration (2 x 107 copies). Values over 0.99 were obtained for the regression coefficient (R2) of the four series (Figure 3).
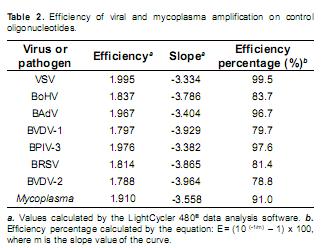
In addition to the observed linear pattern in the standard curves, efficiency percentage for each real time PCR data set was calculated (Table 2). Amplification efficiencies above 78% were calculated for VSV, BoHV-1, BAdV, BPIV-3, BRSV, BVDV-1, BVDV-2 and mycoplasma standard curves, indicating a proper geometrical increase of the copy number along the PCR processes. Maximum efficiency was not obtained for all reactions as we used the same standardized primer and magnesium chloride concentration in the seven specific amplification reactions, looking for establishment of homogeneous amplification conditions which facilitate the analysis procedure.
Detection of viruses and mycoplasma in culture samples
Culture samples. Fourteen samples were obtained and processed to isolate total RNA and DNA. These 14 samples included cellular samples, identified as solid samples (S) and supernatant samples, identified as liquid samples (L), derived from MDBK and HEp-2 cell cultures maintained with reference (GIBCO BRL®) or test (MICROGEN®) FBS (Table 3).
After conversion of total RNA to single-strand cDNA, resultant cDNA and total DNA (DNA) samples were analyzed by conventional PCR amplification for -actin to check for the efficiency of the reverse transcription reaction and for the suitability of these samples to be used as PCR templates.
All total DNA samples from solid samples gave a positive result for Β-actin amplification but liquid samples, corresponding to culture supernatants did not show amplification, except for samples L9 and L10. This must be related with the low amount of nucleic acids present in the culture supernatant (Figure 4). PCR products showed sizes between 100-200 bp, which correspond to the expected sizes (121 bp for bovine derived cells and 133 bp for human derived cells). Size differences between the products were not determined as the resolution of agarose gels is around 10 bp.

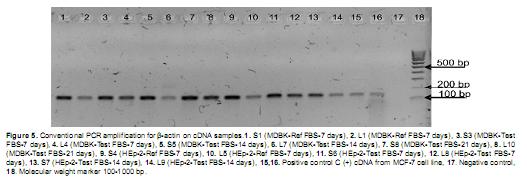
Amplification for Β-actin on unpurified cDNA products was positive for all the tested samples, indicating a successful result for the RT reactions and the suitability of synthesized cDNAs as PCR templates. Obtained PCR products were, as expected, in the 100-200 bp size range (Figure 5).
Detection of bovine viruses. Once samples were confirmed to be useful as PCR templates, viral detection was performed according to cellular sensitivity to viral infection. cDNA samples from MDBK cell line cultures were analyzed for detection of seven pathogenic viruses and Mycoplasma, as this bovine derived cell line is sensitive to the infection of the seven analyzed viruses and also to mycoplasma. On cDNA samples from HEp-2 cell line cultures, PCR detection was only conducted for VSV and BAdV viruses, as this cell line is not sensitive to infection by the other viruses. Each set of samples was analyzed within the same real time PCR run, including negative controls and positive controls corresponding to dilutions of the cloned control oligoA or oligoB as corresponded (Table 4).
Evaluation for viral presence in total DNA samples was conducted only on solid derived samples and the liquid derived samples that showed positive amplification for Β-actin (L9 and L10). BoHV-1, BAdV and Mycoplasma were the only three pathogens analyzed in DNA samples as BoHV-1, BAdV and Mycoplasma have DNA genome, while the other viruses have RNA genome.
Positive controls included in each sample set gave positive amplification, the Ct value obtained for these standards were used by the software to adjust the data set to the standard curve and calculate copy number for each sample. For some of the negative control reactions a Ct value was obtained and a copy number was calculated. These results were considered as noise in the detection, as Ct values for negative controls corresponded to late cycles and the calculated copy number were under the detection limit observed in the standard curves (Table 4).
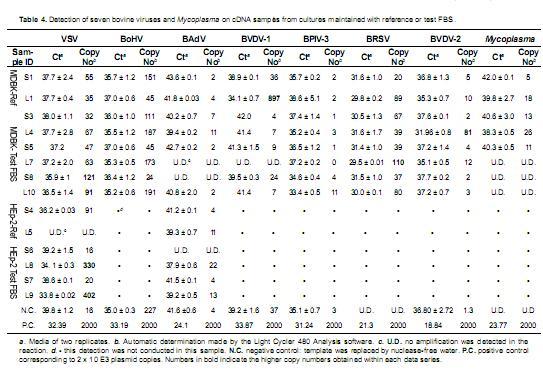
Detection of Vesicular Stomatitis Virus (VSV). A Ct value of 39.8 ± 1.2 was obtained for the negative control and Ct values for the samples were around this value. The calculated copy number for the negative control was 16 viral copies, higher copy number values were obtained for samples derived from MDBK cultures maintained with the reference FBS and test FBS. Among MDBK samples the highest copy number values were obtained for S8 and L10 samples, corresponding to the culture maintained for 21 days with the test-FBS. These values showed to be higher than copy number calculated for cultures maintained 7 and 14 days with the test FBS and 7 days with the reference FBS, nevertheless, 21 days values were in the same order of magnitude. If the VSV were actively present in the culture, a geometrical increase in the detected viral copy number must have occurred along the 21-day follow-up and this viral increase might have been accompanied by cytophatic morphological changes in the cell culture. As this pattern was not observed for MDBK cultures, data are not conclusive for the presence of the VSV. Within the HEp-2 derived samples, high copy numbers were calculated for supernatants from cultures maintained 7 and 14 days with the test FBS. The copy number had a slight increase along thefollow-up period, but not a geometrical increase, in addition no cytophatic effect was observed in these cultures, thus, these data cannot support the presence of the viral infection neither completely discard it in the cultures maintained with the test FBS.
Detection of Bovine Herpes Virus-1 (BoHV-1). Only samples from MDBK cultures were tested for the BoHV-1 presence. A mean Ct value of 35.0 ± 0.3, corresponding to a mean copy number of 227 copies was calculated for the negative control in this data set. Ct values and copy number values calculated for the MDBK cell line cultures maintained with reference-FBS and test-FBS were close to the values for the negative control. None of the calculated copy values was higher than the calculated for the negative control. In addition there was not and increasing pattern in the viral copy number along the culture follow-up period with the test-FBS. These results cannot support the presence of the BoHV-1 in the MDBK cultures treated with the reference or test fetal bovine serum.
BoHV-1 detection was also conducted in the genomic DNA samples that gave a positive amplification for the Β-actin gene. In this set of samples no Ct value was determined for the negative control reactions. In the DNA samples, late Ct values matching with low copy numbers were obtained. In sample S5, from the solid sample of the culture held by 14 days with the test-FBS, a copy number of 190 was obtained, nonetheless, lower copy numbers were obtained for the corresponding S5 cDNA and S8 DNA and cDNA samples, which came from the 21-days culture with test-FBS. Thus, the S5 DNA sample result seems to be a transient increase in viral copy number, but cannot be considered as an indicator of BoHV-1 presence in the MDBK samples treated with the test-FBS.
Detection of the Bovine Adenovirus (BAdV). In this detection a late mean Ct value was obtained for the negative control 41.6 ± 0.6 corresponding to a copy number of 4. Ct values obtained for the samples were late too. These results indicate that the presence of the bovine adenovirus was not detected in the cultures of MDBK and HEp-2 cell lines maintained with the reference-FBS or the test-FBS.
Detection of BAdV in the DNA samples showed amplification with late Ct values for samples obtained from MDBK and HEp-2 cell lines maintained with the reference and test FBS by 7 and 14 days, but no increment on the copy number was observed along the follow-up period. This results togheter with the data obtained for cDNA samples indicate that BAdV was not detected in the treated cultures.
Detection of the Bovine Viral Diarrhea Virus type 1 (BVDV-1). Analysis for BVDV-1 were carried out in MDBK samples. A mean Ct value of 39.2 ± 1.6 was obtained for the negative control and later Ct values were obtained for the samples, except for the L1 sample, supernatant from the culture maintained with the reference-FBS. The copy number for this sample was 897. As no further studies were done with this FBS, this isolated result is not conclusive for the presence of BVDV-1 in the cultures maintained with the reference FBS. No viral presence was detected for MDBK cultured with test-FBS.
Detection of Bovine Parainfluenza Virus 3 (BPIV-3). MDBK cell line cultures were used in this detection. Late Ct value corresponding to low calculated copy numbers were obtained for the negative control, as well as for the samples. No presence of this virus can be deduced from these results (Table 4).
Detection of Bovine Respiratory Syncytial Virus (BRSV). In the analysis for this virus no Ct value was obtained for the negative control. Copy numbers as high as 110 copies were calculated for the supernatant samples, but there was not progressive increase in the copy number detected for samples from 7, 14 and 21-days culture. These results indicate that the high copy number observed for sample L7, the supernatant of the 14-day culture with test-FBS does not represent presence of the respiratory syncytial virus in this cell cultures treated with the test-FBS.
Detection of the Bovine Viral Diarrhea Virus type 2 (BVDV-2). PCR reactions for BVDV-2 were done on MDBK derived samples. Copy number calculated for the negative control and the samples were lower than 10, except for the sample from the supernatant of the culture held for 7 days with the test FBS (Sample L4) and lower values were obtained for the samples of the same culture after 14 and 21 days follow-up, indicating that the L4 sample value is an isolated result and cannot be considered as an indicative for the presence of the tested virus in the MDBK cultures maintained with the test FBS. In addition the calculated copy number was below the linear detection limit established with the standard curve, indicating that this result is not a consistent positive.
Detection of Mycoplasma. cDNA and DNA MDBK derived samples were used in this detection. No amplification was obtained for the negative control reaction and for the DNA samples. For cDNA samples values as high as 26 copy number were obtained for the cDNA samples, for the 7-days culture, but there was no detection in the samples from the 14 and 21-days cultures. These results indicate that Mycoplasma was not present in the MDBK cultures maintained with reference or test FBS.
Discussion
Reports on viral detection by means of real time PCR show that this technique is suitable for the detection of bovine viruses in different biological matrices and demonstrates to be specific and as sensitive as, or more sensitive than the immunological methods like fluorescent antibody test and immunohistochemistry (Timsit et al., 2010).
Specificity of the PCR reaction depends on primer design and can be improved by the use of probes. Primers used in our analysis were tested for specificity by BLAST and no specific probes were used, as detection was made with the ResoLight® DNA-binding dye. Real-time RT-PCR employing the non specific fluorescent SYBR Green-I has been extensively used for bovine viral detection yielding satisfactory results and is also considered as a better option over probes-based amplification, as the result does not rely on the perfect match of the probes, which can be hindered by viral mutations (Kosinova, et al., 2007). Here we replaced the use of SYBR Green-I by ResoLight®, a new non specific DNA-binding fluorescent dye with a higher incorporation frequency, expecting a higher sensitivity. Despite precaution taken in the technical procedures a positive amplification response was obtained for the negative controls. We consider that the highly DNA-incorporation frequency of the Resolight , which accounts for its high sensitivity, might also be responsible for the high noise level detected in these experiments.
The use of cloned viral sequences as positive controls is a resource that allows for generation of standard curves without the use of whole active viruses at the laboratory. This strategy has been used previously and proved to be useful (Kosinova et al., 2007; Young et al., 2006), in our work the constructed control oligonucleotides A and B rendered specific amplification and high linearity standard curves, detecting as low as 2x10E1 pathogen copies for six viruses and mycoplasma and as low as 2x10E2 for BVDV-2. Detection limits for bovine viral diarrhea virus by real-time RT-PCR was established at 1 x 10E1 DNA/µL, copies by other authors, which lies in the same detection level (Young et al., 2006).
For the detection of the seven selected viruses and Mycoplasma, we applied here an enrichment procedure in which susceptible cell lines were cultured for a 21-day period with the test fetal bovine serum MICROGEN®, collecting cellular and supernatant samples each 7 days. This procedure was based on the viral detection test applied to the reference FBS by the manufacturer (APHIS-USA, 1995). The aim of this protocol was the generation of serial data from the same culture, to make a follow- up of the culture. In this way if the presence of a target virus was detected in a sample, this could be compared to the previous and following samples. The projected result was that an increasing copy number along the serial samples taken after 7, 14 and 21 days of culture will account for the presence of the virus.
Prior to real time PCR viral specific amplification, all samples were subjected to Β-actin PCR amplification, to check for the presence of template in samples. Only samples giving a positive amplification in this reaction were analyzed for viral presence, to ensure that negative results will not account for absence of template in the samples. Within the results obtained from the real time PCR reactions there were samples with a high copy number, but these were isolated samples and no increasing pattern was observed when compared within the sample set. A persistent high viral copy number was calculated for the supernatants from the 7 and 14-day follow-up for the VSV, in the HEp- 2 cell line culture maintained with the test FBS, nevertheless the copy number increase was not conclusive for viral presence.
VSV is a highly prevalent virus in Colombia, which has gained space since the eradication of the foot and mouth disease virus (Cajas et al., 2003). This situation explains the possible viral presence detected in the cultures maintained with test-FBS (MICROGEN FBS), even after the filtration by 0.22 µm filters included in the FBS production process. According to this the presence VSV in the Colombian MICROGEN® fetal bovine serum might be considered as one of the highest risks in this product and special attention should be given to the testing of these virus in future analysis for other batches of the product.
Viral analysis carried out in this study did not detect the presence of the pathogenic bovine viruses or Mycoplasma in the tested MICROGEN® fetal bovine serum, except for VSV, which was detected in a low level. The viral detection procedure described here should be included in the quality control analysis of the MICROGEN® fetal bovine serum, to ensure the viral and mycoplasma absence for each batch of the product before its commercialization.
Acknowledgment
This work was supported by the Colombian Administrative Department of Science, Technology and Innovation: Colciencias; contract No 038-2007 (ACAC-Colciencias 179-206).
This work was partially developed at the facilities of the Institute of Biotechnology at the Universidad Nacional de Colombia, Bogotá D.C.
References
1. Animal and Plant Health Inspection Service USDA (US). Code of Federal Regulations, Requirements for ingredients of animal origin. Title 9CFR Part, 113.53. Washington; 1995. [ Links ]
2. Betancur C, Orrego A, González M. Seroepidemiological study of parainfluenza 3 virus in bovines with reproductive failure, from Montería-Colombia. Rev Med Vet 2010; July/ Dec:63-70. [ Links ]
3. Cajas JG, Rojas JA, Almansa JE, Wilches MT, Sánchez C, Acosta O. Comparación de la respuesta inmunológica a las proteínas del virus de la estomatitis vesicular entre bovinos vacunados e infectados de manera natural o inducida. Rev Corpoica 2003; 4:1-5. [ Links ]
4. Freshney RI. Culture of animal cells: a manual of basic technique. 4th ed. Hoboken, (NJ): John Wiley and Sons Inc; 2000. [ Links ]
5. GIBCO Invitrogen Cell Culture. Fetal Bovine Serum Brochure B-066802-r1 0407; 2007. [ Links ]
6. IDT-Integrated DNA Technologies, Inc. PrimerQuest, SciTools. [accesed on July 2009] URL: http://www.idtdna.com/Scitools/ Applications/Primerquest/ [ Links ]
7. Kosinova E, Psikal I, Robesova B, Kovarcik K. Real-time PCR for quantitation of bovine viral diarrhea virus RNA using SYBR Green I fluorimetry. Vet Med 2007; 52:253-261. [ Links ]
8. Nishizawa I, Sato M, Fujihara M, Sato S, Harasawa R.Differential detection of hemotropic Mycoplasma species in cattle by melting curve analysis of PCR products. J Vet Med Sci 2010; 72:77-79. [ Links ]
9. Piedrahita L, Montoya, L Pedraza, F. Bovine Herpes Virus type 1 (BoHV-1) as a possible cause of encephalitis in cattle of the Colombian Magdalena medio region: serological study and epidemiological analysis. Rev Colomb Cienc Pecu 2010; 23:191-198. [ Links ]
10. Rozen S, Skaletsky H. Primer3 on the www for general users and for biologist programmers. In: Krawetz S, Misener S editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa: Humana Press; 2000. p.365-86. [ Links ]
11. Timsit E, Maingourd C, Le Dréan E, Belloc C, Seegers H, Douart A, Assié S. Evaluation of a commercial real-time reverse transcription polymerase chain reaction kit for the diagnosis of bovine respiratory syncytial infection. J Vet Diagn Invest 2010; 22:218-241. [ Links ]
11. Vargas, DS, Jaime J, Vera VJ. Perspectives to control bovine viral diarrhea virus (BVDV). Rev Colomb Cienc Pecu 2009; 22:677-688. [ Links ]
12. Young NJ, Thomas CJ, Collins ME, Brownlie J. Real-time RT-PCR detection of Bovine Viral Diarrhea virus in whole blood using an external RNA reference. J Virol Methods 2006; 138:218-222. [ Links ]

