










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista ION
Print version ISSN 0120-100X
Rev. ion vol.26 no.2 Bucaramanga July/Dec. 2013
on PdxPty bimetallic nanoparticles
Carlos M. Celis-Cornejo; J. Leonardo Gómez-Ballesteros; Sonia A. Giraldo*
Centro de Investigaciones en Catálisis (CICAT), Escuela de Ingeniería Química, Universidad Industrial
de Santander, A.A 678, Bucaramanga, Colombia
*sgiraldo@uis.edu.co
Fecha Recepción: 12 de septiembre de 2013
Fecha Aceptación: 07 de noviembre de 2013
We performed quantum mechanics calculations to elucidate the electronic behavior of Pd-Pt bimetallic nanoparticles, using density functional theory, in response to particle size and stoichiometric composition. Using neutrally charged nanoparticles and the Bader charge analysis, we found that external Pd atoms were positively charged, which agrees with previous XPS observations of supported Pd-Pt nanoparticles. From the calculations, unsupported nanoparticles exhibit an electron transfer from Pd to Pt. This result supports the idea that Pd electron-deficient species are possibly responsible of the hydrogenating function of these catalysts, in the hydrodesulfurization of dibenzothiophene. Additionally, it was found that the particle size does not affect the electronic charge distribution and the stoichiometric composition is the factor that greatly influences this property in nanoparticles.
Keywords: Pd-Pt nanoparticles, Bader charge analysis, DFT, bimetallic clusters.
en nanopartículas bimetálicas de PdxPty
Se llevó a cabo la simulación del comportamiento electrónico de nanopartículas bimetálicas de Pd-Pt mediante cálculos mecanocuánticos, utilizando la teoría del funcional de densidad, con el fin de predecir la influencia en dicho comportamiento, de factores como el tamaño y la composición estequiométrica de la nanopartícula. Al considerar nanopartículas con carga neutra, se encontró, mediante el análisis de cargas de Bader que los átomos externos de Pd presentan una carga positiva, lo cual concuerda con análisis experimentales mediante XPS de catalizadores de Pd-Pt soportados. De las simulaciones, se encontró que las nanopartículas exhiben una transferencia de carga del Pd al Pt. Este resultado da crédito a la idea que especies electrodeficientes de Pd son las responsables de la función hidrogenante, de estos catalizadores, en la hidrodesulfuración del dibenzotiofeno. También se encontró que la influencia del tamaño de la nanopartícula no afecta la distribución electrónica, y que la composición estequiométrica es el factor que más influye en dicha propiedad de las nanopartículas.
Palabras clave: nanopartículas Pd-Pt, análisis de cargas de Bader, DFT, clusters bimetálicos.
Heterogeneous catalysts usually consisting of dispersed metal nanoparticles supported on porous materials such as metal oxides have drawn attention to gain a better understanding of their structure and electronic properties. In contrast to monometallic nanoparticles, bimetallic nanoparticles show a significant improvement in activity, selectivity, stability and prevention of poisoning in oxidation reactions [1,2], hydrogenation [3], and hydrodesulfurization (HDS) [4], owing to synergetic, geometric, and electronic effects [5], and/or mixed sites which can be altered depending on the preparation method [6]. A different and occasionally enhanced behavior, even if the second metal shows poor catalytic activity, has been observed due to the mutual influence of the different neighbor atoms involved in complex metal-metal and metal-support interactions [2,4,7]. Moreover, it is well known that the addition of a second metal species affects the electronic density, and thus the adsorption/desorption phenomena of reagents or intermediate compounds. Depending on the electronic affinity of the second metal added, the catalytic behavior would be enhanced or not [8].
Supported bimetallic nanoparticles such as Pd-Pt/ γ-Al2O3 have been widely investigated as catalysts for the HDS of dibenzothiophenes (DBTs) [4,9,10], and for the oxidation of methane [11]. In the HDS of DBT, a significantly improved selectivity to the hydrogenation pathway of desulfurization was observed at a molar ratio Pd/(Pt+Pd) of 0.8 [4]. The XPS analysis of Pd-Pt/γ-Al2O3 catalysts of different PdXPtY compositions showed an increment in the electron-deficient to metal species relative concentration (Pdδ+/Pd0) at the aforementioned composition. Therefore, it was suggested that Pdδ+ species are responsible for such catalytic behavior [4]. Moreover, similar catalysts used in methane oxidation and prepared through the same co-impregnation method at different compositions, were characterized by XPS by other authors [11]. They found that the enhanced performance against poisoning by SO2, could be related to an electronic effect due to the presence of Pt metallic surface sites at a given composition [11]. A similar trend was observed for Pd-Pt bimetallic catalysts supported on USY Zeolite, for which Pd-Pt ionic interactions were detected by EXAFS [12]. It can thus be noticed that the PdxPty stoichiometric composition is an important variable that might influence the electronic density of the bimetallic nanoparticles. This phenomenon can be addressed using quantum mechanics simulations by means of Density Functional Theory (DFT) studies. This would bring a better insight into the electronic behavior of each metal within the system.
Several Pd-Pt bimetallic systems have been modeled using semi-empirical potentials [13-16]. Previous theoretical studies have clearly shown that Pd tends preferably to occupy the nanoparticle surface [14-16]. The same tendency has been observed in experiments. The phenomenon is best known as surface segregation [15,16]. There are very few quantum mechanical studies available about bimetallic systems. Aprà and Fortunelli studied monometallic Pt clusters of 13, 38 and 55 atoms using DFT [17,18]. They took three different nanoparticle conformations. Namely, icosahedral (Ih), cubo-octahedral (Oh) and truncated decahedral (D5h). For neutral nanoparticles, they found that the most energetically favored structures were icosahedra (Ih) [17,18].
Despite bimetallic systems having an improved catalytic performance compared with monometallic ones, there is not enough detailed information about characterizations of their catalytic active phases [8]. Moreover, there is a lack of theoretical studies aimed to understand electronic effects due to bimetallic interactions, which attempt to correlate these effects with stoichiometric composition of the nanoparticles. Tang et al. [19] performed DFT calculations using plane wave functions for determining electronic densities of transition metal bimetallic clusters, to predict the charge redistribution in the oxygen reduction. For the charge analysis a Bader AIM code was developed [20]. Based on the interpretation of density of states, they could correlate the charge transfer between different metallic species with the Fermi level. Another similar study proposed by Okamura et al. [21], but not as rigorous, studied the effect of replacing the core Pt atom in a pure Pt core-shell structure by a Pd atom and the other way around, over the Mulliken electric charge and the electronic density [21]. They performed DFT calculations using the LANL2DZ basis [21], nevertheless, although this basis is not very reliable for predicting Mulliken charges [22], they obtained reasonable results. It is remarkable that they predicted an electronic transfer from Pd to Pt [21].
As a first step towards the understanding of the electronic behavior of Pd-Pt bimetallic nanoparticles, systems of different particle sizes and compositions with icosahedral shape were considered herein. We wanted thus to address the following question: is the formation of electron-deficient Pdδ+ surface species possible for unsupported Pd-Pt nanoparticles? To answer it, it is fundamental to analyze the distribution of partial atomic charges. Therefore, we carried out quantum mechanic calculations based on DFT using the topological analysis of Bader, to determine how the electronic charge distribution of these bimetallic systems is related to their catalytic activity.
Computational details
Electronic structure calculations were performed using DFT as implemented in the Gaussian03 suite of programs. The UB3PW91 exchange-correlation hybrid functional along with the LANL2DZ Effective Core Potential (ECP) was used. This basis is derived from numerical Dirac-Fock relativistic atomic wave functions, that includes spin-orbital effects [23]; and this ECP was chosen given that relativistic effects heavily influence Pt. As discussed before, core-shell structures with icosahedral point group symmetry of 13 and 55 atoms were considered for different compositions, with interatomic distance of 2.66Å. Pd-Pd, Pd-Pt Interatomic distances were taken the same as Pt-Pt. We considered that this assumption does not affect significantly the results, as we were interested in a trend prediction of the electronic behavior. For a very detailed study, it must be considered that interatomic distances will be different for each case, and they must be obtained by geometry optimizations. The numbers 13 and 55 are magic numbers for the icosahedral shape; these numbers (N) can be easily calculated using the Equation 1, where j is the number of shells.
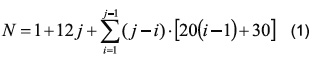
In all cases, neutrally charged nanoparticles were considered, QNET = 0, and spin multiplicity was considered septet for 13-atom and undecatet for 55-atom clusters. QNET calculation is shown in Equation 2. The term QEXTERNAL represents the predicted charge magnitude of each surface atom, and QCENTER is the core atom charge.

Energy optimizations were not performed because of the fact that for 10 group transition metals (Ni, Pd, Pt) self consistent field (SCF) calculations do not converge due to the molecular orbitals lying close to the HOMO which undergo a quasi-degeneracy effect [18,24]. Nonetheless, the latter does not affect the results significantly because our goal was to follow how the stoichiometric composition influences the partial charge distribution, to relate a trend with a catalytic behavior. In fact, many structural possibilities exist at this particle size, and it is well known that for these metallic clusters global minima are low symmetry structures [25]. Perhaps for more realistic conditions the influence of the nanoparticle geometries should be considered as well.
For 13-atom clusters, a total number of 166 structures result from summing all combinations of the possible locations for Pd and Pt in the nanoparticle. For example, consider the composition Pd2Pt11. For this composition 4 possible structures exist (Figure 1). The most energetically favored structures from the 166 possible homotops for 13-atom clusters at each composition were selected. The selection criterion was a single point energy calculation for each structure band. For 55-atom clusters, discrete compositions were considered: Pt55, Pt49Pd6, Pt42Pd13, Pt12Pd43, Pd50Pt5 and Pd55, due to the elevated computational cost at this cluster size. The structures considered were the core-shell arrangement proposed by Cheng et al. [14] for the selected compositions; following the correct building order based on the Many-Body Gupta Potential.
In order to quantify the charge, a Bader topological analysis [26] algorithm as developed by Tang, Sanville and Henkelman [20] was used around each atom. Bader topological analysis was chosen instead of the Mulliken population analysis method, because the latter depends strongly on the selected basis set, leading to misinterpretations of the atomic charges [27]. For this analysis, it is important to obtain a well-defined electron density for every structure. Nonetheless, although it was used the LANL2DZ basis, a comparative study using traditional basis sets against some ECPs showed that Bader topological analysis was the best way for predicting electronic charge, using the Hay-Wadt ECP (LANL2DZ) for transition metals [22]. Thus, for example for Pt, it was there were set in every Bader calculation 18 valence electrons, that LANL2DZ takes into account. The support effect was not taken into account since in previous studies it has been observed that support affects the magnitude of charges but not their distribution, and also that there is a slight tendency of the metallic clusters to become positively charged when the support is hydroxilated [28,29]. Such behavior is reflected in the XPS analysis of supported Pd-Pt nanoparticles owing to differential charging effects.
Charge distribution analysis for 13 atoms clus-ters
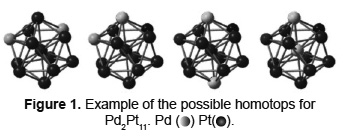
Figure 2 shows the most energetically favored structures of the 166 possible homotops. The absolute energy is listed in atomic units (a.u.), which allows further comparison with the optimal value for each structure. As observed in Figure 2, the Pd atoms preferentially tend to occupy external locations in the cluster instead of the center atom. This proves that the presence of Pd surface atoms is thermodynamically stable, and it is in agreement with experimental evidence on Pd surface segregation [4,5,9,11,13-16,30]. The phenomenon is attributed to differences on sublimation enthalpies and surface energies of the metals. As Pd surface energy is lower than that of Pt, the former tends to segregate to the surface of the bimetallic nanoparticles [14]. Moreover, Pt presents a higher cohesion energy, which is evidenced by a greater Pt-Pt interaction force [13] hence tending to occupy the core of the nanoparticles. The phenomenon has also been documented for supported Pd-Pt nanoparticles; especially, when submitted to highly reducing atmospheres as those present in HDT reactions [4,31]. This not only proves the stability of the nanoparticles proposed herein in realistic catalytic conditions, but also shows that the Pd plays an important role in reactivity. In Figure 2, the conformation noted by C4 shows an appreciable distance between the Pd atoms on the surface. This is due to the binding energy of Pt-Pt bond which is higher than that of both the Pd-Pt and the Pd-Pd bonds [13]. As previously mentioned, this favors an intercalated arrangement of Pd atoms as can be seen in Figure 2. This theoretical prediction is in good agreement with EXAFS evidence on the evolution of the structure of supported Pd-Pt nanoparticles [31].
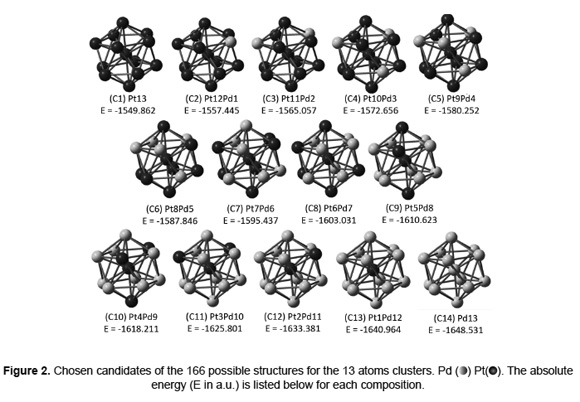
Bader charge analysis was carried out after calculating the electron density of the chosen candidates. Figure 3 shows it as performed for Pd and Pt surface atoms. As can be seen, there is not any Pt surface atom at C13 (Pt1Pd12). The presence of Pdδ+ electron-deficient species due to an electron transfer from Pd to Pt atoms, is predicted. Therein the charge magnitude of the Pt atoms is increased whilst the number of Pd atoms increase.
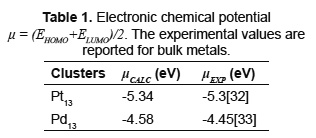
This phenomenon can be explained from an analysis of the electronic structure, calculating the electronic chemical potential (Table 1). The calculated values are close to the experimental data reported in literature for bulk metals [32,33]. The electronic chemical potential of Pd13 cluster is lower than Pt13 cluster. For bimetallic alloys, charge migration occurs from the system with higher chemical potential to the system with lower chemical potential [19]. Now, the following question could be asked: How is the support's effect on the bimetallic nanoparticle charge distribution due to Fermi level shifting? An experimental study based on XANES characterization of Pd-Pt/ASA catalysts shows that the effect of the support composition over the electronic state of the metals within the nanoalloy is negligible [30]. Moreover, in the case of supported Pd-Pt/γ-Al2O3, ab-initio calculations predicted that the hydroxilated support slightly influences the total cluster charge by subtracting electron density from the nanoparticle [28,29]. Hydroxilation of Al2O3 during catalyst preparation involving aqueous impregnation techniques occurs due to electrostatic effects and surface charging of the surface hydroxyls of the oxide [34]. This suggests that positive total charge must be considered in further studies for Pd-Pt/γ-Al2O3 systems, if one were to consider the support effect.
It is important to note that for the C9 (Pt5Pd8) and C10 (Pt4Pd9) conformations (Figure 3), a larger amount of electron-deficient Pdδ+ surface species in the nanoparticles has been observed (Figure 3) with almost the same magnitude. The cited surface compositions are close to a Pd/Pt+Pd atomic ratio of 0.8; for which Baldovino-Medrano et al. made evident a larger amount of electron-deficient Pdδ+ surface species using XPS [4]. Experimental studies on the hydrogenation of aromatic compounds in the presence of sulfur containing compounds have reported a higher reactivity of bimetallic Pd-Pt nanoparticles as compared to monometallic Pd and Pt. It has been proposed that this is due to a charge transfer effect from the Pd to the Pt atoms [35]. It has also been suggested that the lower the electron density of the surface Pd atoms (i.e. a higher value of the Pdδ+/Pd0 ratio) the weaker the metal-sulfur bond; hence the higher the resistance to sulfur poisoning of the catalysts [36].
Charge distribution analysis for 55 atoms clusters
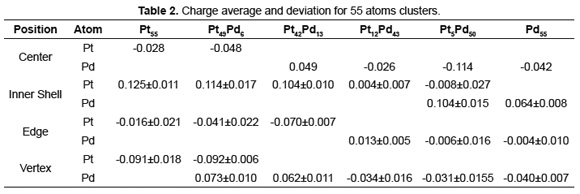
For the 55 atoms clusters, the particle size is around 1.33nm. This is a relevant size considering that in experimental synthesis of bimetallic Pd- Pt supported nanoparticles, using the incipient wetness impregnation method, it was found a particle size distribution between 1.4 and 1.8 nm [30]. Figure 4 shows a Pt 55-atom cluster and the characteristic positions for the external atoms (vertexes and edges). Table 2 summarizes the results of the charge analysis calculations performed for Pd-Pt clusters of 55 atoms. Notice that the charge magnitude is slightly weaker than the one calculated for 13-atom clusters. But the behavior is similar; there is charge migration from Pd to Pt. As seen for both the 13-atom clusters and the 55-atom clusters, the composition is the factor that most influences the charge distribution throughout the nanoparticle.
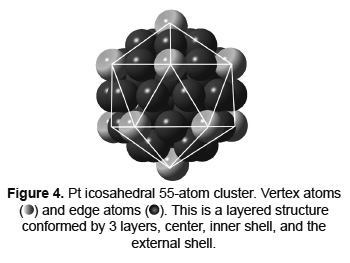
A polarization effect of the shells of the cluster occurs. This is clearly seen in the pure Pt and Pd arrangements, as calculations show but is not well defined in the bimetallic clusters. Despite the polarization effect in the nanoparticles shells over the charge migration on the surface atoms, it is possible to follow the behavior of the Pdδ+ electron-deficient species at this cluster size. The surface Pdδ+ electron-deficient species are presumed to be located on the edges of the icosahedron owing to their higher coordination number as compared to the atoms located on the vertex as for those in Pd rich nanoparticles.
The surface electronic configuration of unsupported Pd-Pt nanoparticles was assessed herein. We found that the presence of Pd in the shell of the nanoparticle was thermodynamically stable and this phenomenon is attributed to a difference in cohesive and surface energies between Pd and Pt. The trend found by the charge analysis of the Pd atoms, confirms the presence of Pdδ+ electron-deficient surface species in bimetallic nanoparticles as a result of a charge transfer process that originates in the electronic chemical potential gradient between Pd and Pt. Furthermore, for 13-atom clusters and for a composition close to 0.8 (Pd/Pd+Pt), a maximum in the concentration of surface Pdδ+ species was found as in agreement with former XPS studies performed on supported Pd-Pt catalysts. Finally we conclude that the stoichiometric composition is the factor that most greatly influences the charge distribution and magnitude in the Pd-Pt bimetallic clusters more than the particle size.
This article is dedicated posthumously to professor Aristóbulo Centeno, who directed this work. We also thank professor Cristian Blanco-Tirado and Dr. V.G. Baldovino-Medrano for his advices and important suggestions. This work was possible due to the financial support of the VIE-UIS in the frame of the project 5462.
[1] Miyake T, Asakawa T. Recently developed catalytic processes with bimetallic catalysts. Appl. Catal. A. 2005;280(1):47-53. [ Links ]
[2] Falletta E, Della Pina C, Rossi M, He Q, Kiely CJ, Hutchings GJ. Enhanced performance of the catalytic conversion of allyl alcohol to 3-hydroxypropionic acid using bimetallic gold catalysts. Faraday Discuss. 2011;152(0):367-79. [ Links ]
[3] Scott RWJ, Wilson OM, Oh S-K, Kenik EA, Crooks RM. Bimetallic Palladium-Gold Dendrimer-Encapsulated Catalysts. J. Am. Chem. Soc. 2004;126(47):15583-91. [ Links ]
[4] Baldovino-Medrano VG, Eloy P, Gaigneaux EM, Giraldo SA, Centeno A. Development of the HYD route of hydrodesulfurization of dibenzothiophenes over Pd-Pt/γ-Al2O3 catalysts. J Catal. 2009;267(2):129-39. [ Links ]
[5] Ferrando R, Jellinek J, Johnston RL. ChemInform Abstract: Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Inform. 2008;39(24):845-910. [ Links ]
[6] Coq B, Figueras F. Bimetallic palladium catalysts: influence of the co-metal on the catalyst performance. J. Mol. Catal. A. 2001;173(1-2):117-34. [ Links ]
[7] Naoki T, Toshiharu T. Preparation, Characterization, and Properties of Bimetallic Nanoparticles. En: Catalysis and Electrocatalysis at Nanoparticle Surfaces. Wieckowski A, Savinova ER, Vayenas CG, Editores. Estados Unidos: CRC Press; 2003. [ Links ]
[8] Dal Santo V, Gallo A, Naldoni A, Guidotti M, Psaro R. Bimetallic heterogeneous catalysts for hydrogen production. Catal. Today. 2012;197(1):190-205. [ Links ]
[9] Baldovino-Medrano VG, Giraldo SA, Centeno A. Highly HYD Selective Pd-Pt/support Hydrotreating Catalysts for the High Pressure Desulfurization of DBT Type Molecules. Catal. Lett. 2009;130(3-4):291-5. [ Links ]
[10] Yoshimura Y, Toba M, Matsui T, Harada M, Ichihashi Y, Bando KK, et al. Active phases and sulfur tolerance of bimetallic Pd-Pt catalysts used for hydrotreatment. Appl. Catal. A. 2007;322(0):152-71. [ Links ]
[11] Corro G, Cano C, Fierro JLG. A study of Pt- Pd/γ-Al2O3 catalysts for methane oxidation resistant to deactivation by sulfur poisoning. J. Mol. Catal. A. 2010;315(1):35-42. [ Links ]
[12] Matsubayashi N, Yasuda H, Imamura M, Yoshimura Y. EXAFS study on Pd-Pt catalyst supported on USY zeolite. Catal. Today. 1998;45(1-4):375-80. [ Links ]
[13] Massen C, Mortimer-Jones TV, Johnston RL. Geometries and segregation properties of platinum-palladium nanoalloy clusters. J. Chem. Soc. Dalton Trans. 2002(23):4375-88. [ Links ]
[14] Cheng D, Huang S, Wang W. Structures of small Pd-Pt bimetallic clusters by Monte Carlo simulation. Chem. Phys. 2006;330(3):423-30. [ Links ]
[15] Rousset JL, Cadrot AM, Cadete Santos Aires FJ, Renouprez AJ, Melinon P, Perez A, et al. Study of bimetallic Pd-Pt clusters in both free and supported phases. J. Chem. Phys. 1995;102:8574-85. [ Links ]
[16] Rousset JL, Cadrot AM, Lianos L, Renouprez AJ. Structure and reactivity of Pd-Pt clusters produced by laser vaporization of bulk alloys. Eur. Phys. J. D. 1999;9(1):425-8. [ Links ]
[17] Aprà E, Fortunelli A. Density-functional study of Pt13 and Pt55 cuboctahedral clusters. J. Mol. Struct. THEOCHEM. 2000;501-502(0):251-9. [ Links ]
[18] Aprà E, Fortunelli A. Density-functional calculations on platinum nanoclusters: Pt13, Pt38, and Pt55. J. Phys. Chem. A. 2003;107(16):2934-42. [ Links ]
[19] Tang W, Henkelman G. Charge redistribution in core-shell nanoparticles to promote oxygen reduction. J. Chem. Phys. 2009;130(19):194504-6. [ Links ]
[20] Tang W, Sanville E, Henkelman G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter. 2009;21(8):084204. [ Links ]
[21] Okumura M, Sakane S, Kitagawa Y, Kawakami T, Toshima N, Yamaguchi K. DFT studies of the characteristics of Pd-Pt core-shell clusters. Res. Chem. Intermed. 2008;34(8-9):737-42. [ Links ]
[22] Anibal S, Fernando R. A comparative study of effective core potential and full electron calculations in Mo compounds. I. An analysis of topological properties of bond charge distribution. J. Comp. Chem. 1994;15(3):313-21. [ Links ]
[23] Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985;82(1):299-310. [ Links ]
[24] Ishiwatari R, Tachikawa M. Unrestricted density functional study on the adsorption of hydrogen molecule on nickel surface. J. Mol.Struct. 2005;735-736(0):383-7. [ Links ]
[25] Radillo-Díaz A, Coronado Y, Pérez LA, Garzón IL. Structural and electronic properties of PtPd and PtNi nanoalloys. Eur. Phys. J. D. 2009;52(1-3):127-30. [ Links ]
[26] Bader RFW. A quantum theory of molecular structure and its applications. Chem Rev. 1991;91(5):893-928. [ Links ]
[27] Ramachandran KI, Deepa G, Namboori K. Computational Chemistry and Molecular Modeling: Principles and Applications. Heidelberg: Springer; 2008. [ Links ]
[28] De Sarkar A, Menon M, Khanra BC. Effect of metal-support interaction on surface segregation in Pd-Pt nanoparticles. Appl Surf Sci. 2001;182(3):394-7. [ Links ]
[29] Hu CH, Chizallet C, Mager-Maury C, Corral-Valero M, Sautet P, Toulhoat H, et al. Modulation of catalyst particle structure upon support hydroxylation: ab-initio insights into Pd13 and Pt13/γ-Al2O3. J. Catal. 2010;274(1):99-110. [ Links ]
[30] Yu Y, Fonfé B, Jentys A, Haller GL, van Veen JAR, Gutiérrez OY, et al. Bimetallic Pt-Pd/ silica-alumina hydrotreating catalysts - Part I: Physicochemical characterization. J. Catal. 2012;292(0):1-12. [ Links ]
[31] Bando KK, Matsui T, Ichihashi Y, Sato K, Tanaka T, Imamura M, et al. In-situ XAFS analysis of dynamic structural change of Pd-Pt nano-particles supported on catalyst surface under sulfidation conditions. Phys. Scr. 2005;2005(T115):828. [ Links ]
[32] Kittel C. Introduction to Solid State Physics. Wiley, editor. USA: John Wiley & Sons, Inc; 2005. [ Links ]
[33] Pearson RG. Chemical Hardness. Weinheim: Wiley-VCH; 1997. [ Links ]
[34] Conţescu C, Vass MI. The effect of pH on the adsorption of palladium (II) complexes on alumina. Appl. Catal. 1987;33(2):259-71. [ Links ]
[35] Bernardi F, Traverse AS, Olivi L, Alves MCM, Morais J. Correlating sulfur reactivity of PtxPd1-x nanoparticles with a bimetallic interaction effect. J. Phys. Chem. C. 2011;115(25):12243- 9. [ Links ]
[36] Seoane XL, L'Argentiere PC, Fígoli NS, Arcoya A. On the deactivation of supported palladium hydrogenation catalysts by thiophene poisoning. Catal. Lett. 1992;16(1):137-48. [ Links ]
