










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.30 no.2 Bogotá Apri./June 2005
Aspectos generales de la eritroenzimopatía más frecuente en el mundo
General aspects of the world's most frequent erithroenzimopathy
Fonseca Dora Janeth, Biol., MSc. Profesor Auxiliar;
Dr. Mateus Heidi Eliana: MSc, Profesor Auxiliar;
Silva Claudia Tamar, Biol., MSc. Profesor Asistente;
Contreras Nora Constanza, Biol., MSc, Instructor Asistente;
Dr. Restrepo Carlos Martín: MSc, PhD(c). Jefe. Unidad de Genética, Instituto de Ciencias Básicas, Universidad del Rosario. Bogotá, D.C.
Conflicto de Intereses: Los autores declaran que no tienen intereses de ningún tipo con las empresas comerciales que puedan beneficiarse de la presente investigación.
Correspondencia: Dra. Dora Fonseca, Calle 63D N° 24-31, Teléfonos: 3474570/60 ext: 266 - e-mail: dfonseca@urosario.edu.co
Recibido: 17/02/05. Aceptado: 19/05/05
Resumen
La deficiencia de la enzima glucosa 6-fosfato deshidrogenasa (G6PD) es la eritroenzimopatía congénita más frecuente en el mundo y se caracteriza por ictericia neonatal (esta enfermedad es asintomática hasta que el portador entra en conctacto con una sustancia de poder oxidante), anemia hemolítica aguda y autolimitada y favismo. Una gran variedad de drogas e infecciones pueden causar anemia hemolítica en las personas con esta deficiencia y las secuelas no hematológicas son bien conocidas. Mediante la caracterización bioquímica establecida por la Organización Mundial de la Salud (OMS), se han documentado a lo largo de diferentes poblaciones en el mundo más de 400 variantes de G6PD; sin embargo, cuatro de ellas se encuentran presentes en la mayoría de las poblaciones: variante A, A-,B y mediterránea. Las técnicas de biología molecular han permitido identificar las mutaciones y/o polimorfismos presentes en el gen que codifica para esta enzima. La distribución de la deficiencia en diferentes poblaciones ha sido investigada exhaustivamente y frecuencias génicas de alrededor de 1,5% se han observado en algunos grupos étnicos, esto ha contribuido no sólo al conocimiento de la enfermedad, sino ha sido relevante en estudios de genética de poblaciones.
Palabras clave: glucosa 6-fosfato deshidrogenasa, diagnóstico, enzimología, genética, epidemiología.
Abstract
Glucose 6 phosphate dehydrogenase (G6PD) deficiency is the most frequent congenital enzymophaty around the world. It is characterized by neonatal jaundice, haemolytic anemia and favism. A diverse variety of drugs and infections can induce haemolytic anaemia in people with this illness. With a standardized biochemical characterization established by the World Health Organization it has been possible to identify more than 400 variants of G6PD around the world. However, only four major variants, A, A-, B and Mediterranean variants, are found in most populations. With molecular biology techniques it is possible to identify mutations and polymorphism in the G6PD gen. The distribution of the G6PD deficiency has been widely investigated around the world and the gene frequency of the most common variant in some ethnics groups is around 0,5. This new knowledge has contributed to increase understanding of this illness and its importance in population genetic studies.
Key-words: glucose-6-phosphate dehydrogenase, diagnosis, enzymology, genetics, epidemiology.
Introducción
La deficiencia de la G6PD es una eritroenzimopatía causada por el bloqueo de la vía enzimática de la hexosa monofosfato que lleva al acúmulo de peróxido de hidrógeno causando daño oxidativo al eritrocito. Fue descrita por primera vez por Alving y colaboradores cuando investigaban la reacción inusual que ocurría en personas de raza negra luego de la administración de primaquina, para el tratamiento de la malaria (1), históricamente se ha atribuido el fallecimiento de Pitágoras, a su negativa a ingresar a una plantación de habas, dada su condición de favismo, posiblemente secundaria a la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD). Posteriormente, se ha relacionado este defecto con cuadros clínicos que incluyen hemólisis intravascular masiva como una reacción a múltiples drogas y químicos, hemólisis luego de la ingestión de habas (favismo) e ictericia neonatal severa con kernicterus. Alrededor de 400 millones de personas en el mundo padecen esta enfermedad, y así la alta frecuencia de esta entidad determina que esta eritroenzimopatía puede considerarse como un problema de salud pública, especialmente en regiones con alta incidencia de malaria o con acervos genéticos procedentes de estas regiones. Muchos países han incluido a la deficiencia de G6PD en programas de tamizaje genético neonatal, dado que la formación de bilirrubina no conjugada puede producir ictericia nuclear, una de las principales causas de retardo mental y muerte en los neonatos.
Características bioquímicas
Estructura de la enzima
La glucosa-6-fosfato deshidrogenasa es una enzima muy antigua en la evolución, ya que se encuentra en todos los organismos vivientes, desde levaduras y protozoos a plantas y animales. En los mamíferos es citoplasmática y se encuentra en todas las células del cuerpo, pero su deficiencia se manifiesta más en los glóbulos rojos posiblemente por tener éstos una larga vida sin núcleo y porque contienen proteasas que degradan la enzima mutante más que las proteasas de otros tejidos (2). El monómero de la G6PD consta de 515 aminoácidos con un peso molecular de 59256 daltons. La G6PD del hígado y de los leucocitos, presentan diferencias debidas a modificaciones postraduccionales en el extremo N-terminal. La enzima activa consiste de subunidades idénticas que forman dímeros y tetrámeros, la proporción de las dos formas depende del pH (3), contiene un sitio de unión a nicotinamida-adenina-dinucleotido-fosfato (NADP) (4), y así la agregación de los monómeros inactivos a la forma de dímeros catabólicamente activos requiere de la presencia de NADP (5). El NADP se une a la enzima, como un componente estructural y como sustrato para la reacción (4). Se ha sugerido que los sitios de unión de esta coenzima son los aminoácidos 386 y 387, que corresponden a lisina y arginina, esta observación ha sido soportada por estudios en mutantes en quienes se ha evidenciado pérdida de actividad ante la ausencia de estos sitios (6).
La G6PD cataliza el paso de entrada de glucosa 6-fosfato (G6P) en la vía de las pentosa fosfato, específicamente en la de la hexosa monofosfato, reacción que produce oxidación de la glucosa-6-fosfato a 6-fosfogluconolactona, reduciendo NADP a NADPH. En el glóbulo rojo, este paso anaeróbico en el metabolismo de la glucosa es la única fuente de NADP reducido (NADPH), el cual es requerido para la acción normal de la metahemoglobina reductasa y el mantenimiento de un nivel adecuado de glutation reducido (4). La glutation peroxidasa remueve el peróxido del eritrocito (7); el glutation reducido sirve como sustrato para esta enzima y debido a que NADPH es esencial para la reducción del glutation oxidado, es un factor esencial en las cadenas de reacción que defienden al glóbulo rojo del peróxido (8). Los glóbulos rojos son una fuente particularmente rica de catalasa, pero esta enzima es relativamente ineficiente en la remoción de bajos niveles de peróxido. Además, la catalasa tiene la habilidad para unir fuertemente a NADPH (9) y la forma inactiva es reactivada por NADPH. Por tanto, la actividad de la vía de las hexosas sirve para remover el peróxido no sólo a través de la acción de la glutatión peroxidasa si no también activando las catalasas (10). Por tanto, ambas enzimas desempeñan un papel muy importante y sirven como un mecanismo de base la una para la otra.
La deficiencia de la enzima provoca un daño oxidativo irreversible en el eritrocito causando su muerte. La vida media de esta enzima es de 60 días y refleja paso a paso la edad del glóbulo rojo, ya que éste es incapaz de formar nuevas moléculas proteicas y es por esto que el reticulocito tiene cinco veces más actividad enzimática que los glóbulos senescentes (11).
Las características bioquímicas de la enzima han sido referenciadas a través de estudios de movilidad electroforética, de la determinación de la constante de Michaelis (Km), pH óptimo, y termoestabilidad y así se han caracterizado más de 400 variantes de G6PD, algunas asociadas a las formas más severas de la enfermedad.
Genética
Herencia e inactivación del X
Incluso antes de que fuera descrito el defecto básico, ya había sido descrito un modo de herencia ligado al X para la "sensibilidad a la primaquina". Los árboles genealógicos de familias afroamericanas mostraban que el defecto era transmitido principalmente de madres a hijos (12). Con la identificación gracias a estudios de medición de la actividad enzimática, movilidad electroforética, etc. (13) del defecto básico, como la deficiencia de la G6PD, se confirmó ligamiento al cromosoma X. El gen de la G6PD está localizado en la región terminal del brazo largo del cromosoma X (Xq28), cercano al gen del factor VIII de la coagulación.
Dado el tipo de herencia de la deficiencia de G6PD, los hombres son normales o deficientes, las mujeres pueden ser normales, heterocigotas u homocigotas; es bastante difícil establecer esta diferenciación basándose únicamente en la expresión fenotípica. Las mujeres heterocigotas tienen una copia del gen que sintetiza la G6PD normal y otra copia que produce la variante de la enzima; así, estas pacientes tienen dos poblaciones de eritrocitos, una normal y otra con deficiencia de G6PD; este hecho se ha explicado por la inactivación al azar de uno de los dos cromosomas X en las mujeres (14).
Gen G6PD
El gen fue secuenciado y clonado por dos grupos independientes Pérsico y Takizawa en 1986 (15, 16). El gen está constituido por 13 exones y tiene una longitud de alrededor de 20 kb. Los exones tienen tamaños que varían entre 38 y 236 pb; el primer exón tiene una secuencia no codificante y los intrones son pequeños, excepto el intrón 2 (11 kb). La longitud total del gen es de 2,4 kb y el RN Am consta de 2269 nucleótidos. En el extremo 5' del gen existe una isla rica en CpG (dinucleótidos histidina-guanina). La demetilación diferencial de algunos CpG se asocia con la expresión del gen en el cromosoma X activo (17).
Variantes asociadas con deficiencia de G6PD
En 1967, un comité de la OMS propuso los procedimientos bioquímicos estandarizados para caracterizar las variantes de G6PD, teniendo como parámetros de evaluación: la actividad enzimática, el km para la enzima y el NADP, estabilidad al calor, eficiencia de utilización de G6P, movilidad electroforérica y pH óptimo. La caracterización ha permitido la identificación de 442 variantes bioquímicas distintas, y cerca de 100 de ellas son polimórficas en varias poblaciones humanas (18-23). Las variantes polimórficas son aquellas que se encuentran con alta frecuencia en algunas poblaciones y representan polimorfismos balanceados. La variantes polimórficas mejor conocidas son la G6PD Mediterránea, la variante Africana (G6PD A-) y las variantes orientales. Generalmente cada población tiene sus propias características mutacionales (18). Las variantes esporádicas han sido también reportadas en muchas poblaciones y son caracterizadas por anemia hemolítica crónica no esferocítica; en algunos países no todas las variantes analizadas desde el punto de vista bioquímico han sido caracterizadas a nivel molecular, es más, en algunas ocasiones se ha encontrado variantes que bioquímicamente son diferentes pero que portan la misma mutación (24), observación que ha sido atribuida a la utilización de métodos de identificación diferentes a los establecidos por la OMS: las variantes han sido agrupadas en cinco clases, basados en la actividad enzimática residual y el los síntomas clínicos: La Clase I está asociada con anemia crónica no esferocítica y es secundaria a mutaciones puntuales, las cuales están confinadas a dos áreas cerca al extremo carboxiterminal de la enzima, en la región entre los aminoácidos 362 y 446, en donde se encuentran los sitios de unión a NADP o NADPH y el sitio de unión a la glucosa 6-fosfato. Se han descrito cerca de 97 variantes, siendo apenas una polimórfica, por ejemplo: variantes Andaloris, Campinas y Sumaré. La Clase II con una deficiencia enzimática severa (menor del 10%) y anemia hemolítica aguda, con 122 variantes, siendo 37 polimórficas, por ejemplo las variantes Mediterránea y Unión. La Clase III con deficiencia moderada (10-60%); 103 variantes, siendo 22 polimórficas (variantes Africana o A-, Canton y Seattle). La Clase IV con actividad enzimática normal o ligeramente disminuida (60-100% de actividad); 52 variantes, con 12 polimórficas (variante A). La Clase V con un aumento en la actividad enzimática (>150%), dos variantes, ninguna polimórfica (variante Verona) (23). Posteriormente se han descrito mutaciones que producen variantes de Clase I, localizadas en un agrupamiento dentro del exón 10, en una región involucrada en la dimerización de la proteína, mientras que la mayoría de las mutaciones leves se localizan en el extremo amino de la molécula (25) (Tabla 1). Como los defectos intragénicos han sido identificados, muchas variantes que se pensaban diferentes han resultado con una secuencia idéntica luego del análisis.
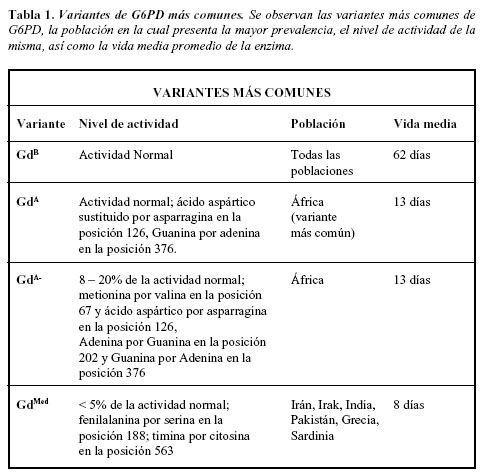
Mutaciones asociadas a G6PD
Una de las características más importantes del gen de G6PD es la gran cantidad de mutaciones que lo afectan (Tabla 2), produciendo enzimas con actividad o cantidad anormal. Gran número de las mutaciones corresponden a sustituciones de una sola base y generalmente se encuentran dispersas en todo el gen, excepto en los exones 3 y 13. Las mutaciones puntuales que resultan en variantes de Clase I están confinadas a áreas relacionadas con el sitio de unión de NADP o NADPH. La mutación Africana es la G6PDA- y es debida a una transición A por G en el nucleótido 376. Muchos individuos muestran la presencia de una segunda mutación G por A en el nucleótido 202; la principal mutación mediterránea corresponde a una transición C por T en el nucleótido 563; en la población oriental es común encontrar la variante Canton localizada en el nucleótido 1376, aún se ha documentado una considerable heterogeneidad mutacional en asiáticos (18, 26).
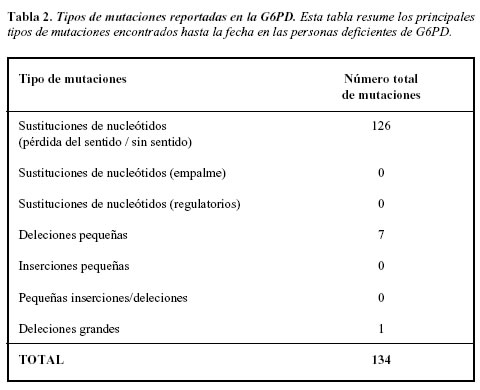
A la fecha se han descrito más de 123 mutaciones en el gen de la G6PD y algunas de ellas evaluadas mediante diferentes estrategias moleculares como microarreglos se han relacionado a poblaciones muy específicas, lo cual hace necesario establecer el espectro mutacional propio de cada país, tendiente a identificar de una manera rápida y sensible las mutaciones relevantes en cada población (24).
La deficiencia de la G6PD como un polimorfismo balanceado
Cuando un gen que tiene potencial para producir alguna alteración adquiere una alta frecuencia en algunas poblaciones, es necesario asumir que en estas poblaciones también confiere una ventaja selectiva. Cuando se llega a un balance entre la ventaja y la desventaja conferidas por el gen, éste se designa como polimorfismo balanceado. Uno de los más estudiados es la mutación para la anemia de células falciformes y la ventaja conferida por este gen es la resistencia a la malaria falciparum. La mortalidad causada por la malaria en algunas partes del mundo es tan alta, que un gran número de rasgos genéticos que defienden contra la infección han sido involucrados, y varios de los polimorfismos que afectan el eritrocito al parecer han alcanzado altas incidencias por esta razón (27). La distribución geográfica de la deficiencia de G6PD ha llevado a varios autores a sugerir que la deficiencia de esta enzima es uno de los polimorfismos que confiere resistencia a la infección por malaria (13, 28, 29), además se ha documentado una disminución de la parasitemia dentro de los pacientes deficientes de G6PD, comparados con individuos normales (13) y un menor número de parásitos encontrados en las células deficientes comparadas con las células con G6PD suficiente en los pacientes heterocigotos (30).
Incidencia y distribución geográfica
La deficiencia de G6PD es la enzimopatía conocida más común, con una incidencia promedio a nivel mundial de alrededor del 1% estando afectados por esta deficiencia más de 400 millones de personas (90% hombres) (29) distribuidos principalmente en las regiones del Mediterráneo, Medio Oriente, India, Indochina, sur de China así como en África (31).
De todas las variantes de G6PD con actividad deficiente, las que muestran mayor importancia por su frecuencia son: la variante Africana o A-, que ocurre comúnmente en descendientes de este continente, así como del sur de Italia, España, Portugal y la península arábiga; la variante Mediterránea que se encuentra usualmente en italianos de Sardinia y Sicilia y en griegos, judíos orientales, árabes y persas; la variante Canton frecuente en el sur de China, la variante Seattle que tiene frecuencias polimórficas en Sardinia, Grecia, el sur de Italia y Estados Unidos y finalmente la variante Unión descrita en chinos y en el sur de Italia.
En Suramérica se han realizado estudios en Brasil, encontrando más frecuentemente las variantes Africana y Mediterránea (32); en México donde luego del análisis de 1938 personas observando sólo tres variantes polimórficas A-202A/376G, A-376G/968C y Seatle; en Cuba se ha encontrado una presencia de la variante Africana en el 7% de la población (33, 34).
En la Tabla 3 se presenta una revisión de los estudios realizados en Suramérica, donde se demuestra una clara distribución étnica, reportando las variantes B, A, A- y Mediterránea como las más comunes en la población de raza negra o caucásica, mientras que la mayoría de los amerindios presentaban la variante B.
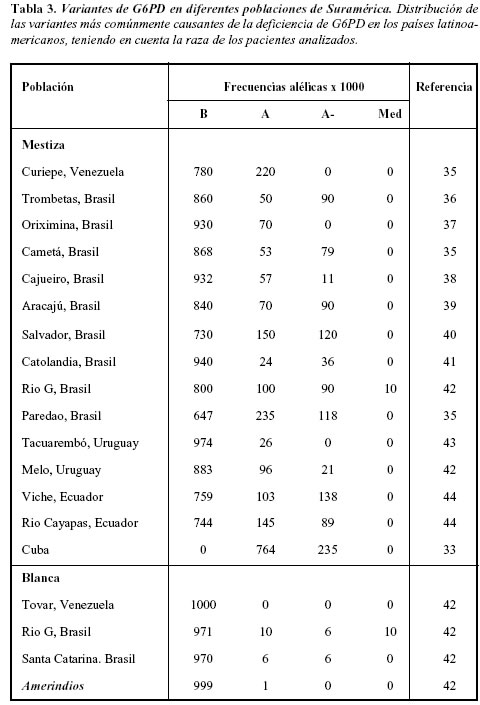
En Colombia, no se han reportado en la literatura estudios acerca de la prevalencia de esta entidad o de sus variantes.
Diagnóstico
Un gran número de métodos bioquímicos cuali y cuantitativos además de análisis moleculares son disponibles para el diagnóstico de la deficiencia de G6PD; dentro de las evaluaciones cualitativas, los métodos utilizados son la determinación de actividad de G6PD sobre papel de filtro mediante técnicas fluorescentes, y el método de indofenol-diclorofenol (DPIP). En la medición sobre manchas de papel de filtro, el NADPH generado en la reacción por personas no deficientes de G6PD es detectado visualmente bajo luz UV, mientras que en el método DPIP se detecta la presencia de G6PD por decoloración del marcador en un tiempo específico (45-47). La cuantificación de la enzima involucra la medición espectrofotométrica de la reducción de NADP a NADPH en presencia de G6P y hemolizado (23). La detección de la deficiencia de G6PD durante estados agudos de hemólisis puede generar dificultades ya que los reticulocitos son muy ricos en la enzima. Con el desarrollo de las técnicas de biología molecular se han utilizado diferentes metodologías que han permitido conocer las causas moleculares que llevan a la deficiencia de G6PD, dentro de ellas están la PCR alelo específica, análisis de polimorfismo conformacional de cadena sencilla (SSCP), identificación de polimorfismos de restricción de longitud variable (RLFP) y microarreglos (2, 24, 48-50).
Manifestaciones clínicas
La mayoría de las personas que cursan con deficiencia de G6PD son usualmente asintomáticas y sólo se manifiesta la enfermedad cuando éstos ingieren drogas o químicos que desencadenan la hemólisis masiva intravascular. La expresión clínica entonces resulta de la interacción de las propiedades moleculares de cada variante de G6PD con factores exógenos y posiblemente factores genéticos adicionales específicos para determinadas poblaciones. Se han descrito diferentes síndromes clínicos asociados a la deficiencia de esta enzima que incluyen:
Hemólisis inducida por drogas. Clásicamente, luego de la ingestión de ciertos agentes, el paciente va a desarrollar fiebre, orina de coloración negra, ictericia y anemia. En ocasiones esta hemólisis se puede complicar con un cuadro de necrosis tubular y coagulación intravascular diseminada. La lista de las drogas que producen hemólisis en los pacientes deficientes de G6PD se enumeran en la Tabla 4.

Hemólisis inducida por infección. La infección es probablemente la causa más común de hemólisis en los pacientes con deficiencia de G6PD. La severidad y las consecuencias clínicas de la hemólisis están influenciadas por numerosos factores que incluyen la administración simultánea de drogas oxidantes, los niveles de hemoglobina previos, la función hepática y la edad. El mecanismo por el cual la infección induce la hemólisis no es conocido y continúa siendo uno de los temas de estudio (23).
Favismo. La presentación de cuadro de hemólisis aguda luego de la ingestión de habas, ha sido reconocida desde la antigüedad (51), presentando los pacientes un cuadro clínico similar al inducido por fármacos, que se desencadena dentro de las 24 y 48 horas siguientes a la ingesta de habas. Sin embargo, no todos los pacientes con deficiencia de G6PD presentan hemólisis luego de la ingesta de habas (52). Los compuestos vicina e isouramilo, abundantes en las habas, se piensa que son los causantes de la respuesta hemolítica (23).
Los pacientes afectados también pueden desarrollar ictericia neonatal con kernicterus, la cual ocurre clásicamente entre los días 4 y 7 posnatales, es decir, en una forma más tardía que la icteria neonatal secundaria a incompatibilidad ABO o Rh. En las personas deficientes de G6PD esta no es el resultado de la hemólisis, sino el resultado del compromiso hepático, debido a la deficiencia de esta enzima en el hígado (23).
Anemia hemolítica crónica no esferocítica. las variantes de Clase I se caracterizan por este hallazgo, debido al grado tan severo de deficiencia enzimática. La hemólisis es sólo parcialmente intravascular y se puede acompañar de cálculos biliares y esplecnomegalia. Sin embargo, existe una gran variabilidad en las manifestaciones asociadas a este tipo de anemia crónica.
Referencias
1. Carson PE, Flanagan CL, Ickes CE, Alving AS. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science 1956; 124: 484-5. [ Links ]
2. Dal Borgo P, Silva R, Cavieres M. Dos nuevas mutaciones de glucosa 6 fosfato deshidrogenasa, G6PD Santiago y G6PD Calvo Mackenna. Rev Chil Pediatr 2000; 71: 419-22. [ Links ]
3. Wrigley NG, Heatrher JV, Bonsignore A, DeFlora A. Human erythrocyte glucose 6-phosphate dehydrogenase: Electron microscope studies on structure and interconversion of tetramers, dimers and monomers. J Mol Biol 1972; 68: 483-499 [ Links ]
4. De Flora A, Morelli A, Guilano F. Human erythrocyte glucose 6-phosphate dehydrogenase. Content of bound coenzyme. Biochem Biophys Res Commun 1974; 59: 406-13. [ Links ]
5. Kirkman HN, Hendrickson EM. Glucose 6-phosphate dehydrogenase from human eryhrocytes. II. Subactive states of the enzyme form normal persons. J Biol Chem 1962; 237: 2371-6. [ Links ]
6. Hirono A, Kuhl W, Gelbart T, Forman L, Fairbanks VF, Beutler E. Identification of the binding domain for NADP+ of human glucose-6-phosphate dehydrogenase by sequence analysis of mutants. Proc Natl Acad Sci USA 1989; 86: 10015-7. [ Links ]
7. Gaetani GF, Galiano S, Canepa L, Ferraris AM, Kirkman HN. Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood 1989; 73: 334-9. [ Links ]
8. Srivastava SK, Beutler E. Glutathione metabolism of the erytrocyte. The enzymic cleavage of glutathione-haemoglobin preparations by glutathione reductase. Biochem J 1970; 119: 353-7. [ Links ]
9. Kirkamn HN, Gaetani GF. Catalase: A tetrameric enzyme with four tightly bound molecules of NADPH. Proc Natl AcadSci USA 1984; 81: 4343-8. [ Links ]
10. Kirkman HN, Galiano S, Gaetani GF. The function of catalase-bound NADPH. J Biol Chem 1987; 262: 660-7. [ Links ]
11. Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Seminars of Hematology 1990; 27: 1-40. [ Links ]
12. Childs B, Zinkham W, Browne EA, Kimbro EL, Tobert JV. A genetic study of a defect in glutathione metabolism of the erythrocyte. Johns Hopkins Med J 1958; 102: 21- 37. [ Links ]
13. Boyer SH, Porter IH, Weilbacher RG. Electrophoretic heterogeneity of glucose-6-phosphate dehydrogenase and its relationship to enzyme deficiency in man. Proc Natl Acad Sci USA 1962; 48: 1868-1876. [ Links ]
14. Beutler E, Yeh M, Fairbanks VF. The normal human female as a mosaic of X-chromosome activity: Studies using the gene for G6PD deficiency as a marker. Proc Natl Acad Sci USA 1962; 48: 9-16. [ Links ]
15. Persico MG, Viglietto G; Martino G, Toniolo D, Paonessa G, Moscatelli C, et al. Isolation of human glucose-6-phosphate dehydrogenase (G6PD) cDNA clones: Primary structure of the protein and unusual 5' non-coding region. Nucleic Acids Res 1986;14: 2511-20. [ Links ]
16. Takizawa T; Huang IY, Ikuta T, Yoshida A. Human glucose-6-phosphate dehydrogenase: primary structure and cDNA cloning. Proc Natl Acad Sci USA 1986; 83:4157-61. [ Links ]
17. Toniolo D, Martini G, Migeon BR, Dono R. Expresión of the G6PD locus on the human X chromosome is associated with demethylation of three CpG islands within 100kb of DNA. EMBO J 1988; 7: 401-10. [ Links ]
18. Mohanty D, Mukherjee MB, Colah RB. Glucose-6-phosphate dehydrogenase deficiency in India. Indian J Pediatr 2004;71:525-9. [ Links ]
19. Beutler E. G6PD deficiency. Blood 1994; 84:3613-36. [ Links ]
20. Frigerio R, Sole G, Lovicu M, Passiu G. Molecular and biochemical data on some G6PD variants from southern Sardinia. Haematologica 1994; 79:319-21. [ Links ]
21. Calabró V, Mason PJ, Filosa S, Civitelli D, Cittadella R, Tagarelli A, et al. Genetic heterogeneity of glucose-6-phosphate dehydrogenase deficiency revealed by single-strand conformation and sequence analysis. Am J Hum Genet 1993; 52:527-36. [ Links ]
22. Viglietto G, Montanaro V, Calabro V, Vallano D, D'Urso M, Persico MG, et al. Common glucose-6-phosphate dehydrogenase (G6PD) variants from the Italian population: biochemical and molecular characterization. Ann Hum Genet 1990; 54:1-15. [ Links ]
23. Beutler E, Yoshida A. Genetic variation of glucose-6-phosphate dehydrogenase: a catalog and future prospects. Medicine(Baltimore) 1988; 67:311-34. [ Links ]
24. Bang-Ce, Hongqiong L, Zhensong L. Rapid detection of common Chinese glucose-6-phisphate dehydrogenase (G6PD) mutations by microarray-based assay. Am J Hematol 2004; 76: 405-12. [ Links ]
25. Costa E, Cabeda JM, Vieira E, Pinto R, Pereira SA, Ferraz L, Santos R, Barbot J. Glucose-6-phosphate dehydrogenase aveiro: a de novo mutation associated with chronic nonspherocytic hemolytic anemia. Blood 2000; 95:1499-501. [ Links ]
26. Stevens DJ, Wanachiwanawin W, Mason PJ, Vulliamy TJ, Luzzatto L. G6PD Canton a common deficient variant in South East Asia caused by a 459 Arg - Leu mutation. Nucleic Acids Res 1990; 18:7190-202. [ Links ]
27. Nagel RL, Roth EF Jr. Malaria and red cell genetic defects. Blood 1989; 74: 1213-21. [ Links ]
28. Greene LS. G6PD deficiency as protection against falciparum malaria: An epidemiologic critique of population and experimental studies. Yearbook Phys Anthropol 1993; 17 (suppl 36): 153-60. [ Links ]
29. Wajcman H, Galacteros F. Glucose 6-phosphate dehydrogenase deficiency: a protection against malaria and a risk for hemolytic accidents. C R Biol 2004; 327: 711-20. [ Links ]
30. Luzzatto L, Usanga EA, Reddy S. Glucose-6-phosphate dehydrogenase deficient red cells. Resistance to infection by malarial parasites. Science 1969; 164: 839-42. [ Links ]
31. Chan TK, Todd D. Characteristics and distribution of glucose-6-phosphate dehydrogenase-deficient variants in South China. Am J Hum Genet 1972; 24: 475-84. [ Links ]
32. Compri MB, Saad ST, Ramalho AS. Genetico-epidemiological and molecular investigation of G-6-PD deficiency in a Brazilian community. Cad. Saude Publica 2000; 16: 335-42. [ Links ]
33. Estrada M, Gutierrez A, Palacios B, Perez G, Rovira A, Vives JL. Estudio bioquímico y molecular de la glucosa-6-fosfato deshidrogenasa en Cuba. Revista Cubana de Hematología, Inmunología y Hemoterapia 1995; 11: 115-120. [ Links ]
34. Gonzalez R, Ballester JM, Estrada M, Lima F, Martinez G, Wade M, et al. A study of the genetical structure of the Cuban population: Red cell and serum biochemical markers. Am J Hum Genet 1976; 28: 585-96. [ Links ]
35. Bortolini MC, Weimer TA, Franco MH, Salzano FM, Layrisse Z, Schneider H, et al. Genetic studies in three South American Black populations. Gene Geogr 1992; 6: 116. [ Links ]
36. Schneider H, Guerreiro JF, Santos SE, Weimer TA, Schneider MPC, Salzano FM. Isolate breakdown in Amazonia The Blacks of the Trombetas River. Rev Bras Genet 1987; 10: 565 74. [ Links ]
37. Santos SEB, Guerreiro JF, Salzano FM, Weimer TA, Hutz MH, Franco MHLP. Mobility, blood genetic traits and race mixture in the Amazonian population of Oriximina. Rev Bras Genet 1987;10:74559. [ Links ]
38. Bortolini MC, Weimer TA, Zago MA, Da Silva WA Jr, Guerra DC, Salzano FM, et al. Protein and Protein and hypervariable tandem repeat diversity in eight African-derived South American populations: inferred relationships do not coincide. Hum Biol 1998; 70: 443-61. [ Links ]
39. Conceiçao MM, Salzano FM, Franco MHLP, Weimer TA, Krieger H. Demography, genetics, and race admixture in Aracaju, Brazil. Rev Bras Genet 1987; 10: 31331. [ Links ]
40. Bortolini MC, Weimer TA, Salzano FM, Moura LB, Silva MCBO. Genetic structure of two Afro-Brazilian populations. Int J Anthr 1997; 12: 516. [ Links ]
41. Weimer TA, Tavares-Neto J, Franco MHLP, Hutz MH, Salzano FM, Kubo RR, Rosa RTD, Friedrisch J, Prata A. Genetic aspects of Schistosoma mansoni infection severity. Rev Bras Genet 1991;14: 62330. [ Links ]
42. Weimer TA, Salzanoa FM, Westwoodb B, Beutlerb E. G6PD Variants in Three South American Ethnic Groups: Population Distribution and Description of Two New Mutations. Hum Hered 1998; 48: 9296. [ Links ]
43. Sans M, Alvarez I, Bentancor N, Abilleira D, Bengochea M, Sosa M, Toledo R, et al. Blood protein genetic markers in a northeastern Uruguayan population. Rev Bras Genet 1995;18: 317320. [ Links ]
44. Martinez-Labarga C, Rickards O, Scacchi R, Corbo RM, Biondi G, Pen JA, et al. Genetic Population Structure of Two African-Ecuadorian Communities of Esmeraldas. Am J Phys Anthrop 1999; 109:15974. [ Links ]
45. Beutler E, Mitchell M. Special modifications of the fluorescent screening method for glucose6-phosphate dehydrogenase deficiency. Blood 1968; 32: 816-8. [ Links ]
46. Bernstein RE. A rapid screening dye test for the detection of glucose-6-phosphate dehydrogenase deficiency in red cells. Nature 1962; 194: 192-3. [ Links ]
47. Betke K, Beutler E, Brewer GJ, Kirkman HN, Luzzato L, Ramot B, et al. Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. Report of a WHO Scientific Group. World Health Organ Tech Rep Ser. 1967; 366:1-53. [ Links ]
48. Xu W, Westwood B, Bartsocas CS, Malcorra-Azpiazu JJ, Indrak K, Beutler E. Glucose 6 phosphate dehydrogenase mutations and haplotypes in various ethnic groups. Blood 1995; 85: 257-63. [ Links ]
49. Du CS, Ren X, Chen L, Jiang W, He Y, Yang M. Detection of the most common G6PD gene mutations in Chinese Using amplification refractory mutation system. Human Heredity 1999; 49: 133-8. [ Links ]
50. Maffi D, Pasquino MT, Caprari P, Caforio MP, Cianciulli P, Sorrentino F, et al. Identification of G6PD Mediterranean mutation by amplification refractory mutation system. Clin Chim Acta 2002; 321: 43-7. [ Links ]
51. Fermi C, Martinetti P. Studio sul favismo. Ann Igiene Sper 1905; 15: 75-86. [ Links ]
52. Siniscalco M, Bernini L, Latte B, Motulsky AG. Favism and thalassaemia in Sardinia and their relationship to malaria. Nature 1961.190: 1179-83. [ Links ]

